[DeNovo Protocols → Step-by-Step → DeNovo Protocols]

Figure 5: Submenu to follow a de novo analysis protocol in Step-by-step mode
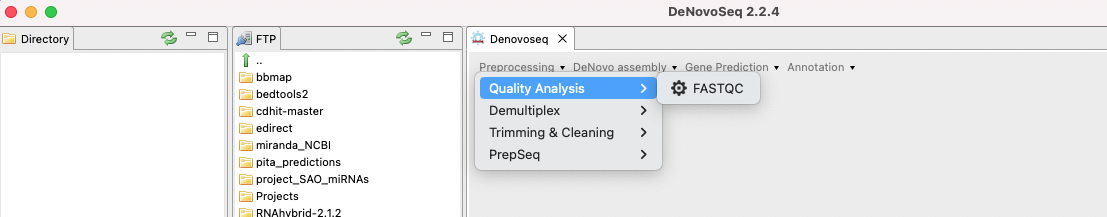
Figure 6: Preprocessing options enabled in DeNovoSeq.
- Quality Analysis: FastQC
FastQC (Andrews 2016) provides a simple way to perform quality checks on raw sequence data. It provides a modular set of analyses that can indicate whether your data contain potential artifacts that require “cleaning” before beginning any analyses. Upon performing a FastQC quality check, you will obtain a complete sequence quality report provide hints on what form of filtering and processing your sample requires if any. For example, overrepresented sequences that correspond to the adapters used during sequencing may need to be removed from your fastq files. This will be shown in the “overrepresented sequences” section of the FastQC report. Further details on FastQC can be found in the FastQC manual at https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
To run FastQC go to [Preprocessing → Quality Analysis → FastQC] and follow Fig. 7
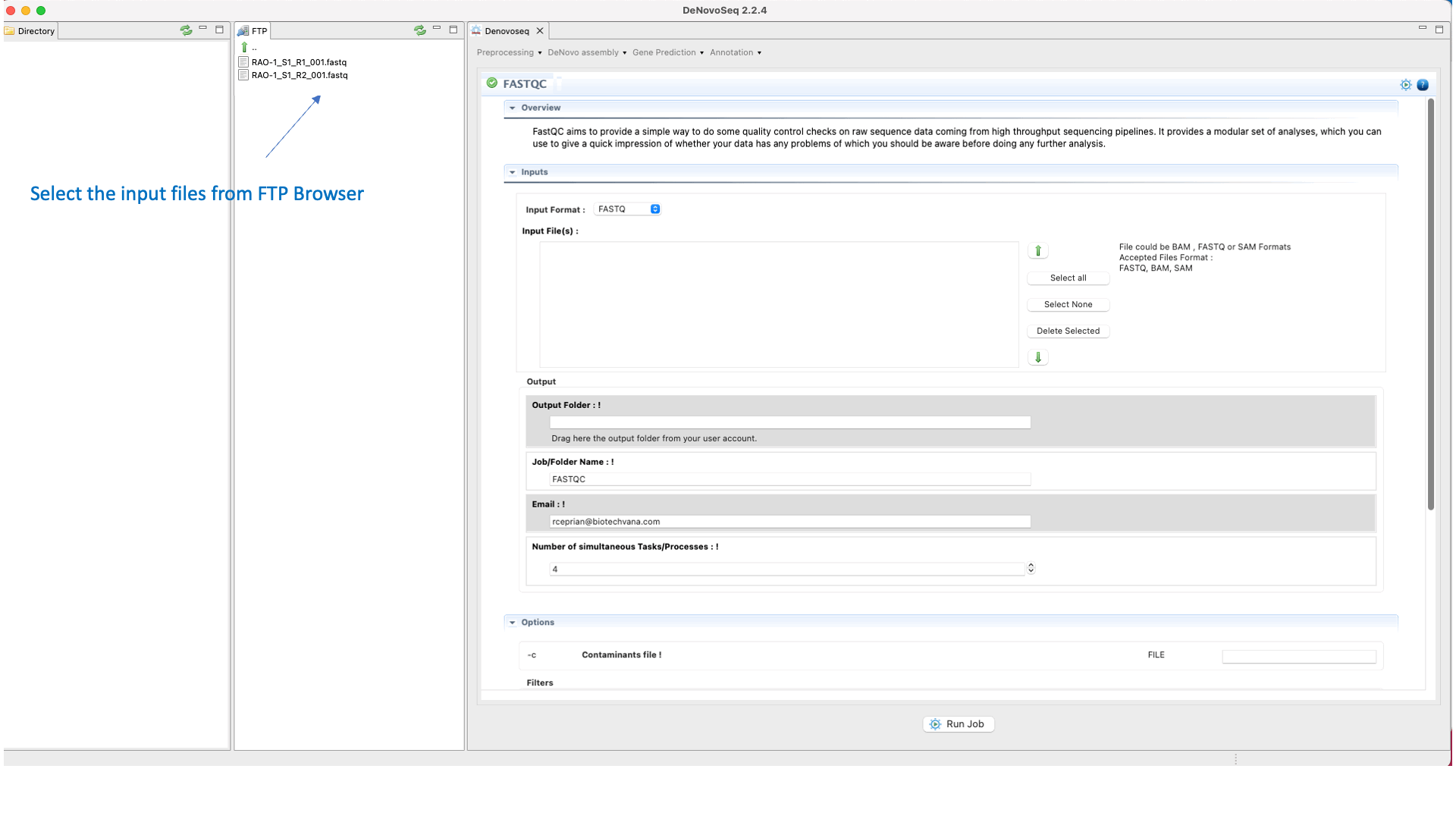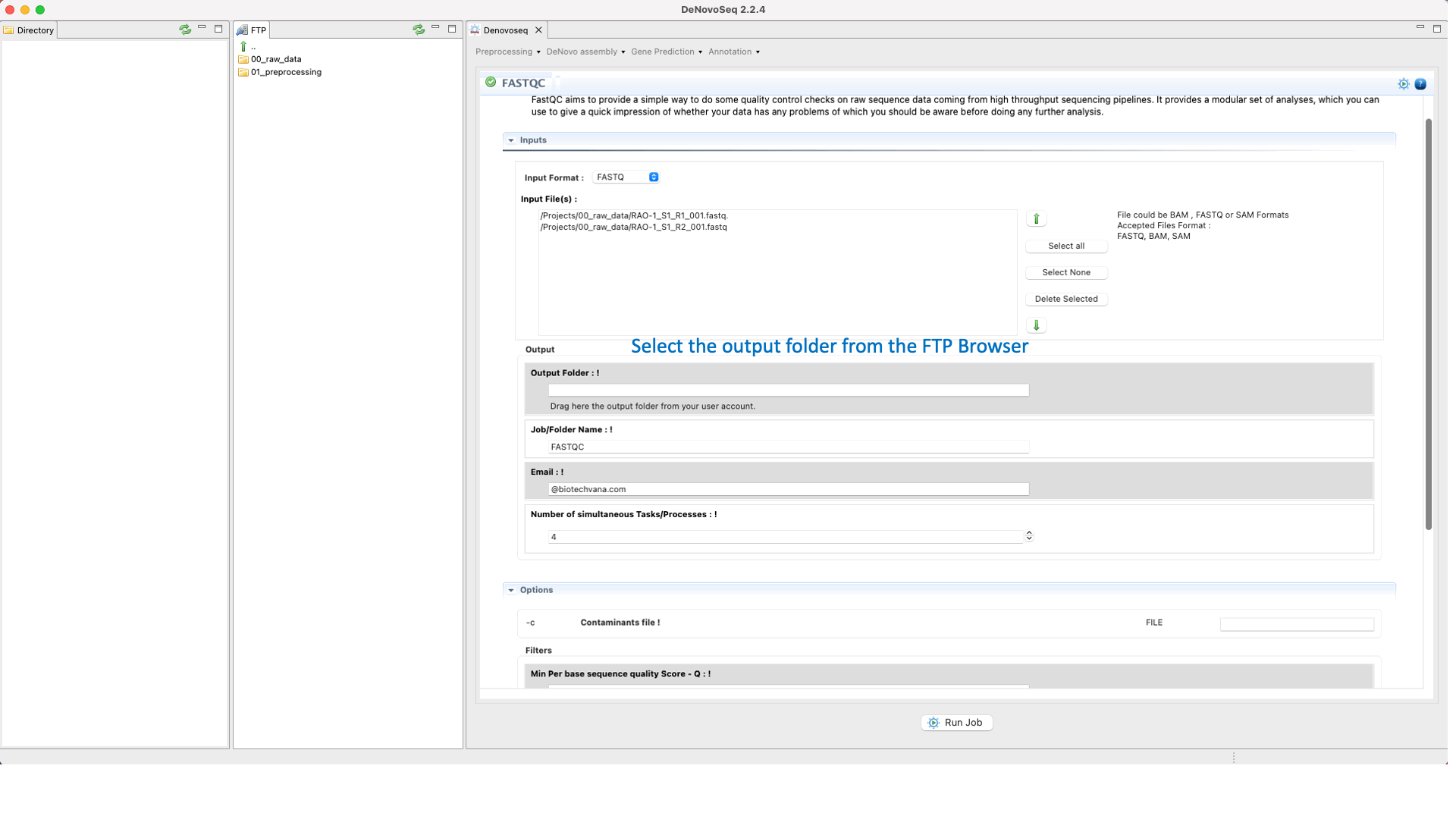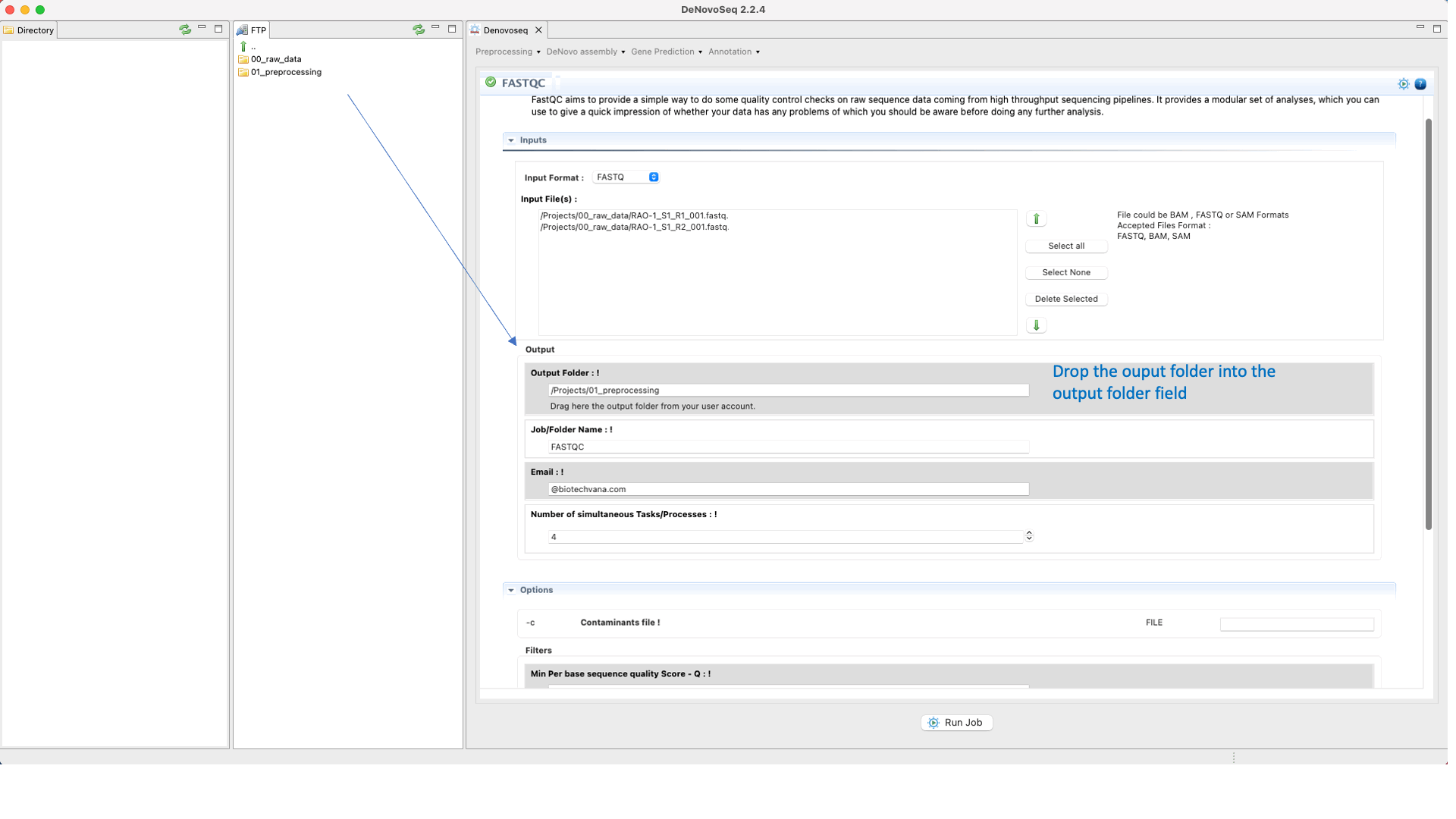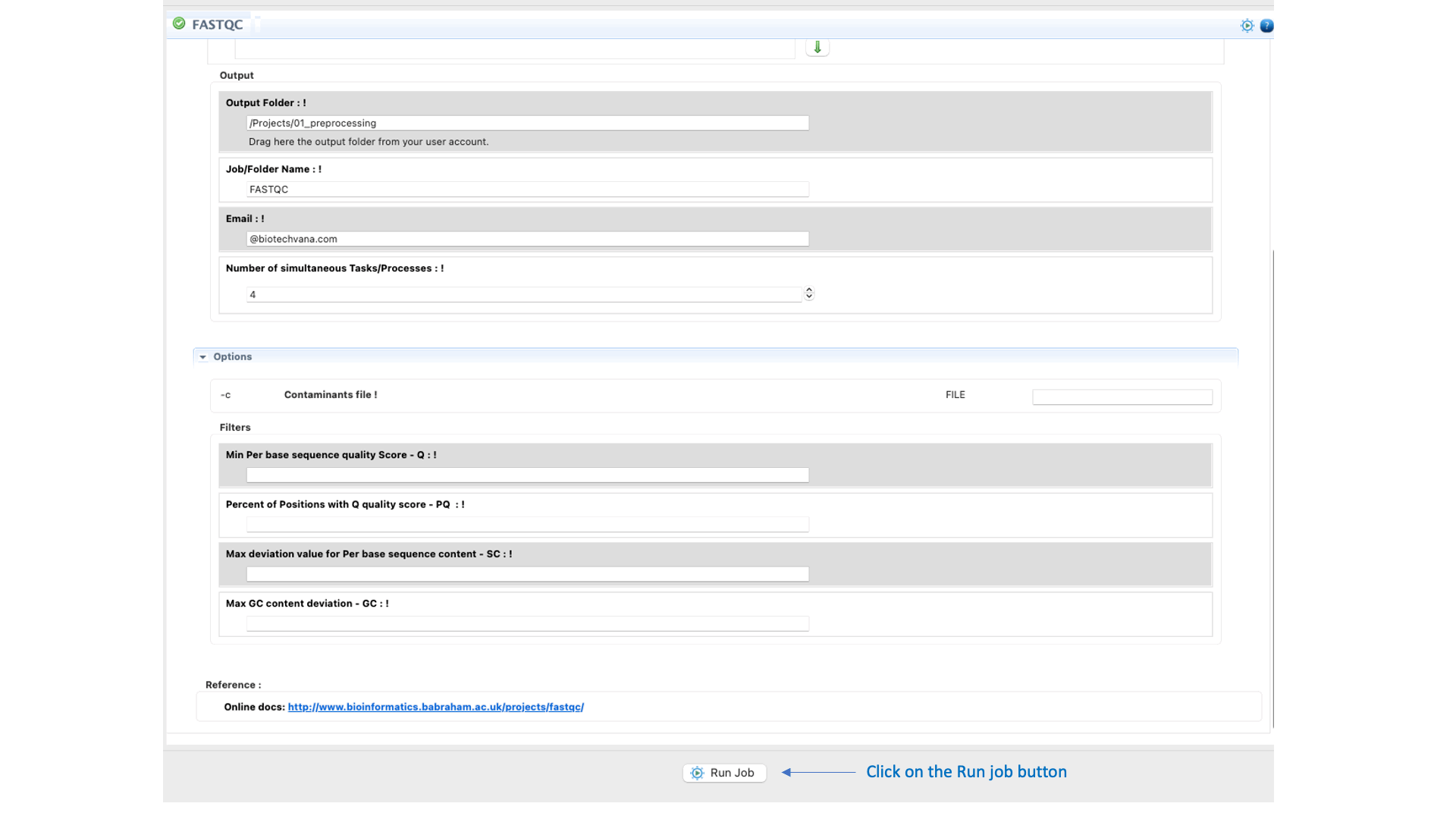
Figure 7: Using the GPRO interface for FastQC.
- Demultiplex: FastqMidCleaner
FastqMidCleaner sorts and splits sequencing reads from fastq files into separate files according to predefined molecular identifiers (MIDs).
To run FastqMidCleaner go to [Preprocessing → Demultiplex → FastqMidCleaner] and follow Fig. 8.
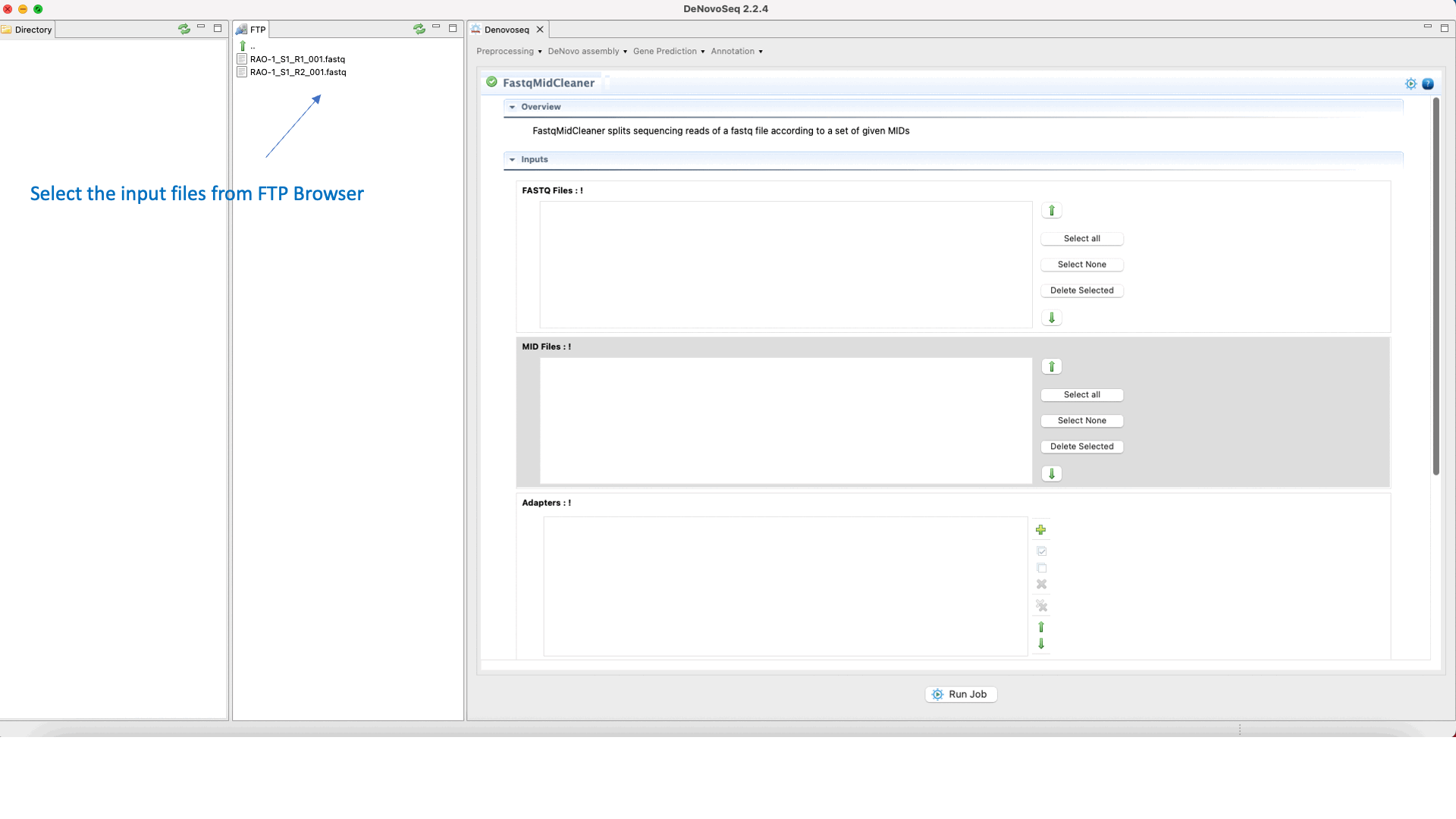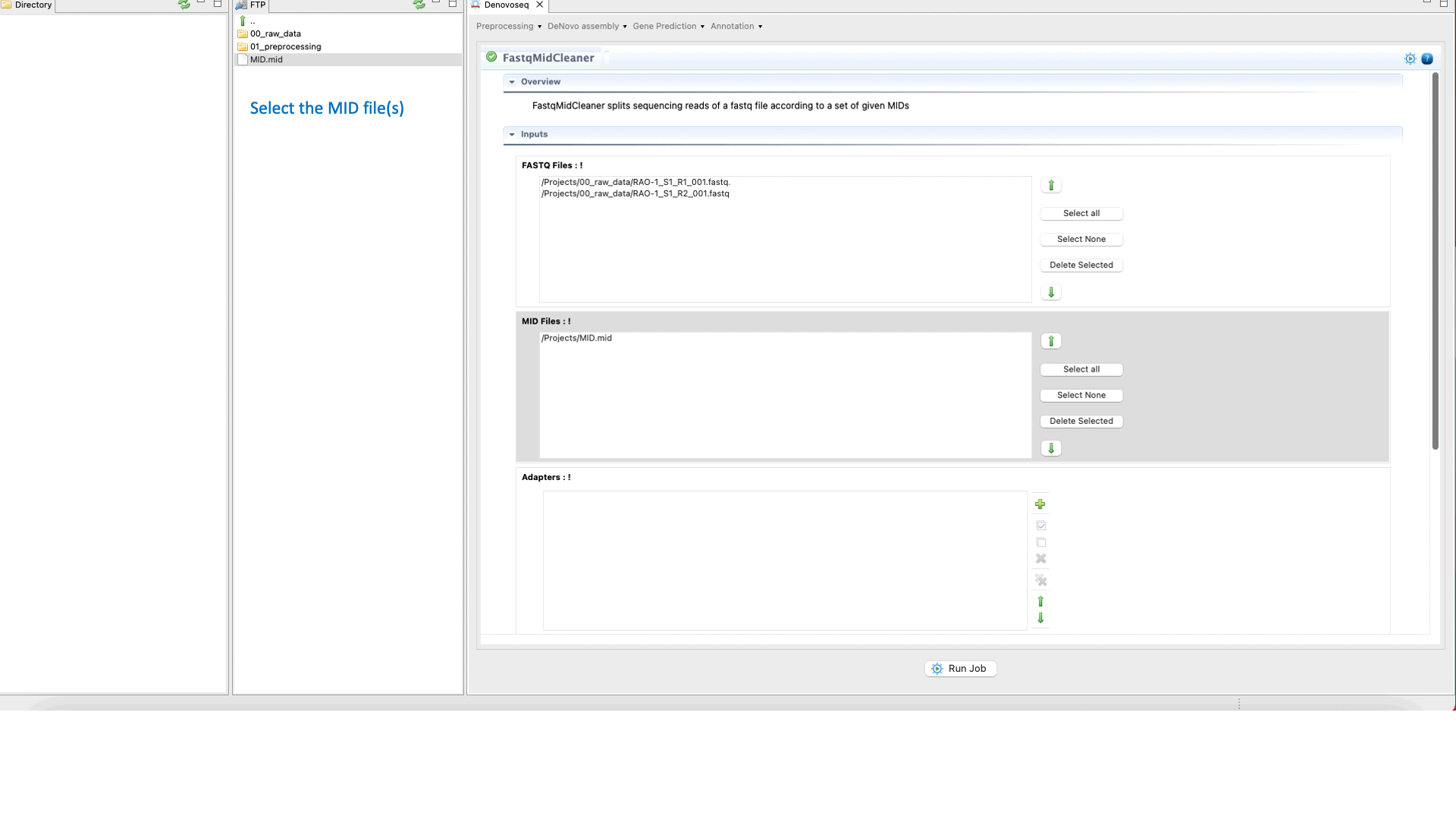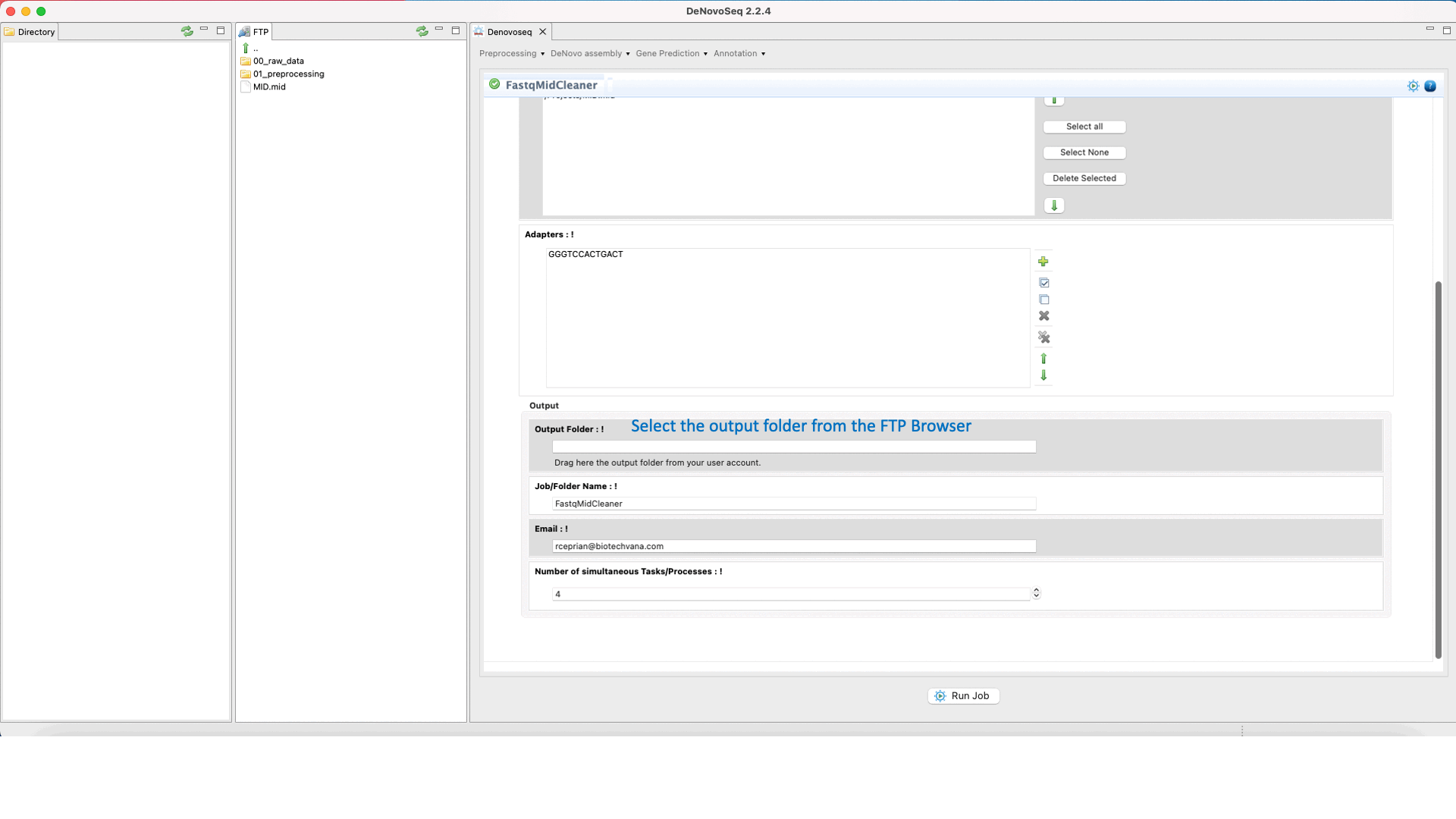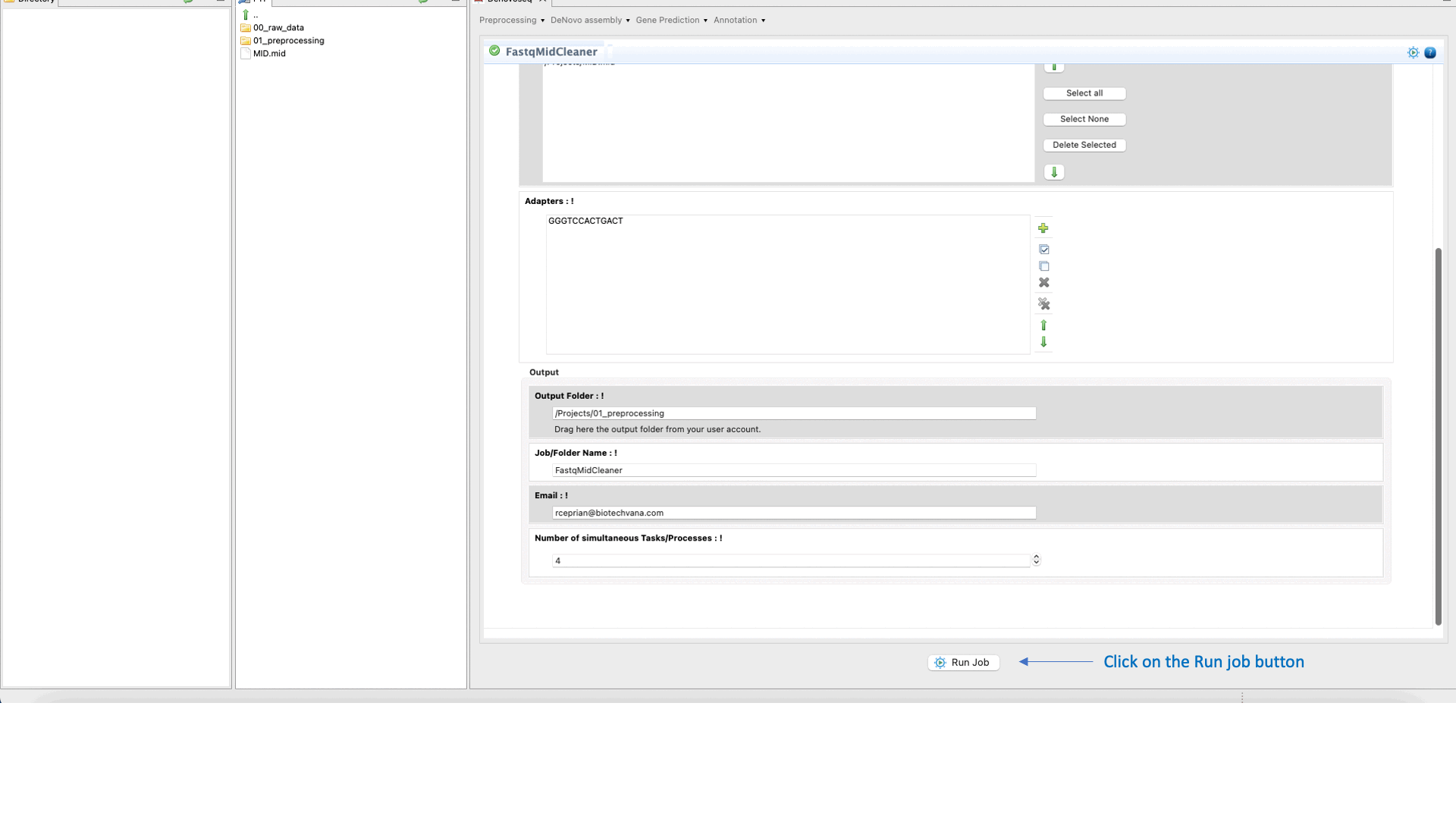
Figure 8: Using the GPRO interface for FastQMidCleaner.
Cutadapt (Martin 2011) finds and removes adapter sequences, primers, poly-A tails and other types of unwanted sequences from your sequencing reads. For more information on Cutadapt see the Cutadapt manual at https://cutadapt.readthedocs.io/en/stable/guide.html
To run Cutadapt go to [Preprocessing → Trimming & Cleaning → Cutadapt] and follow Fig. 9.
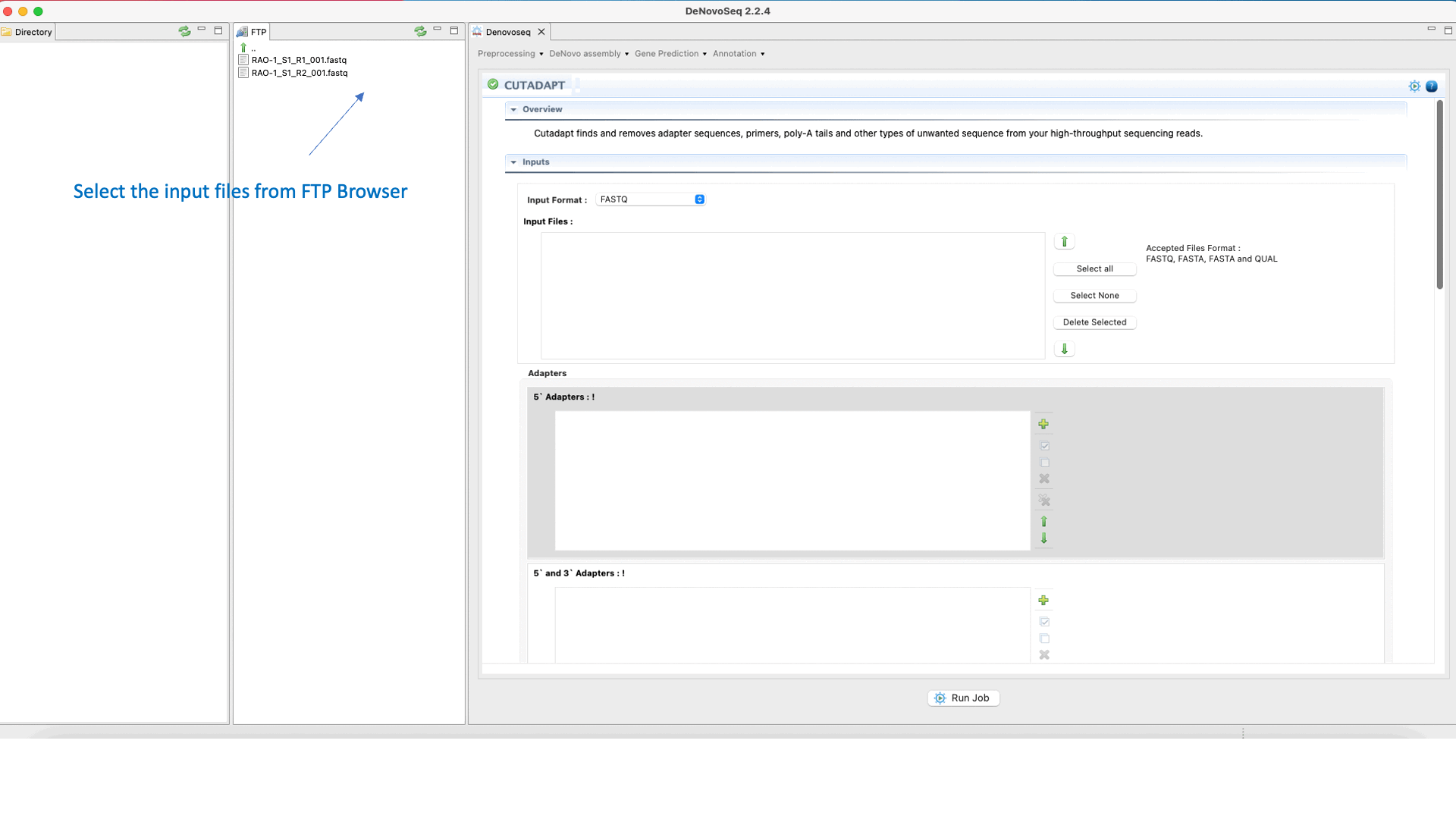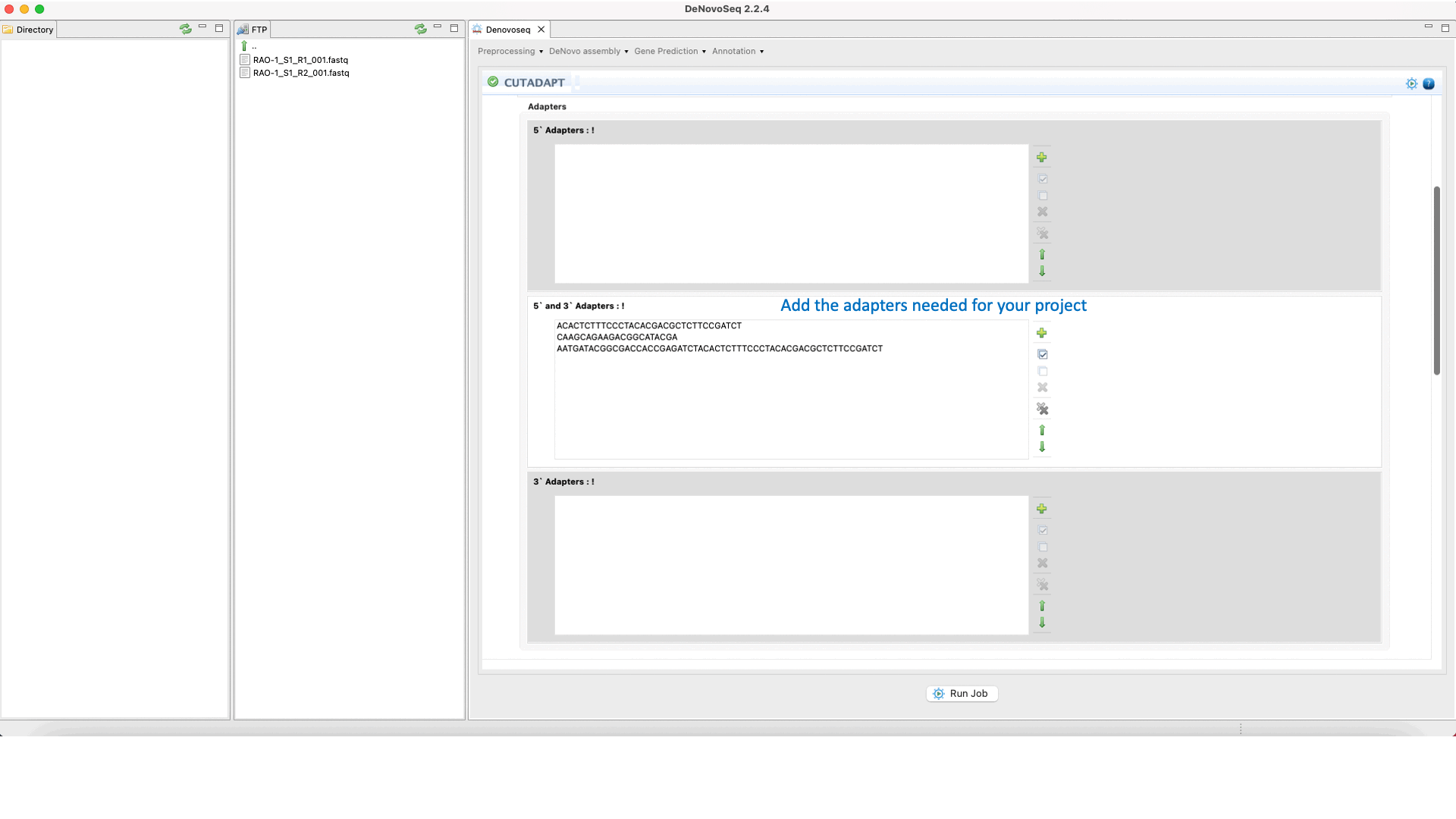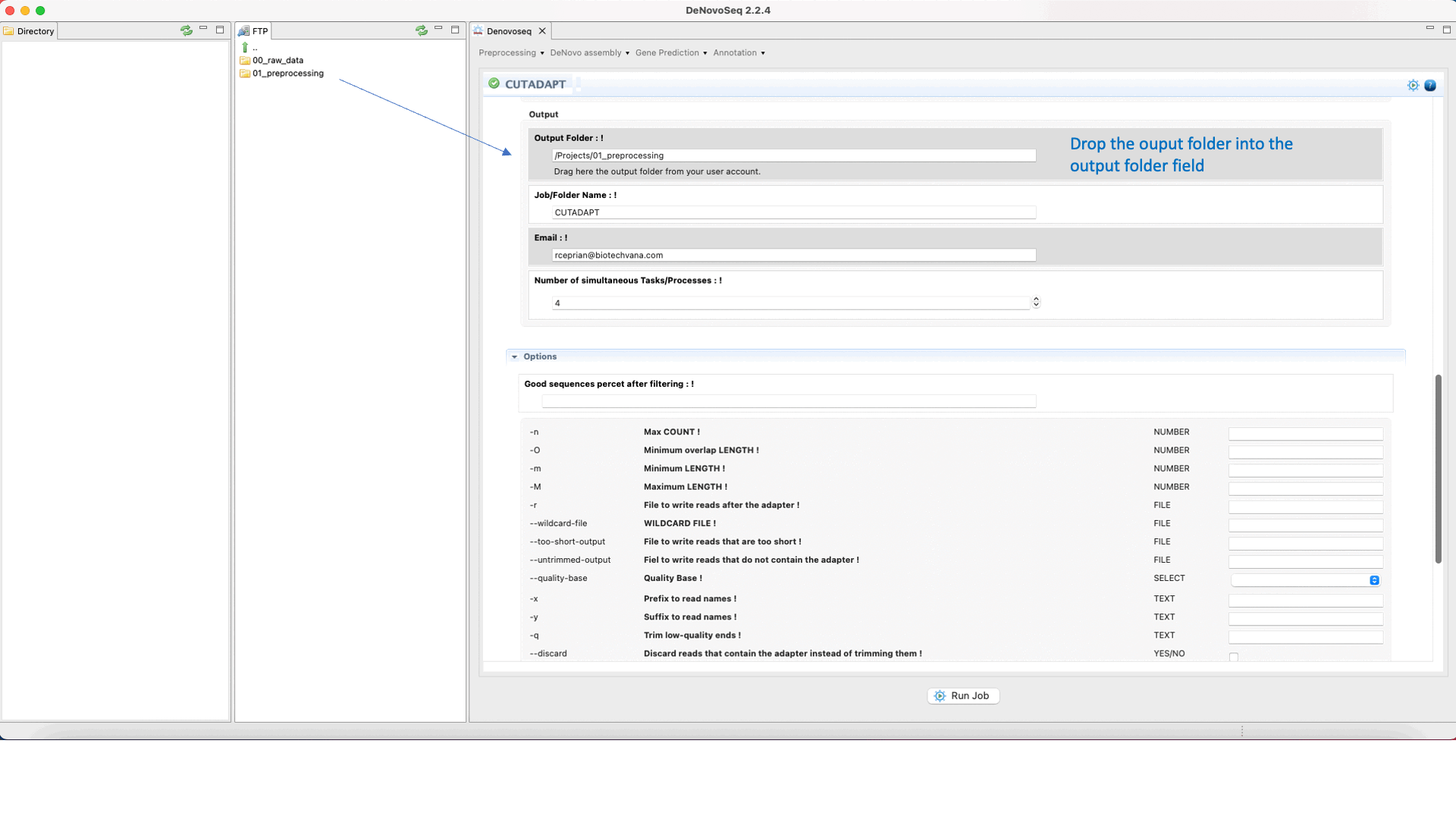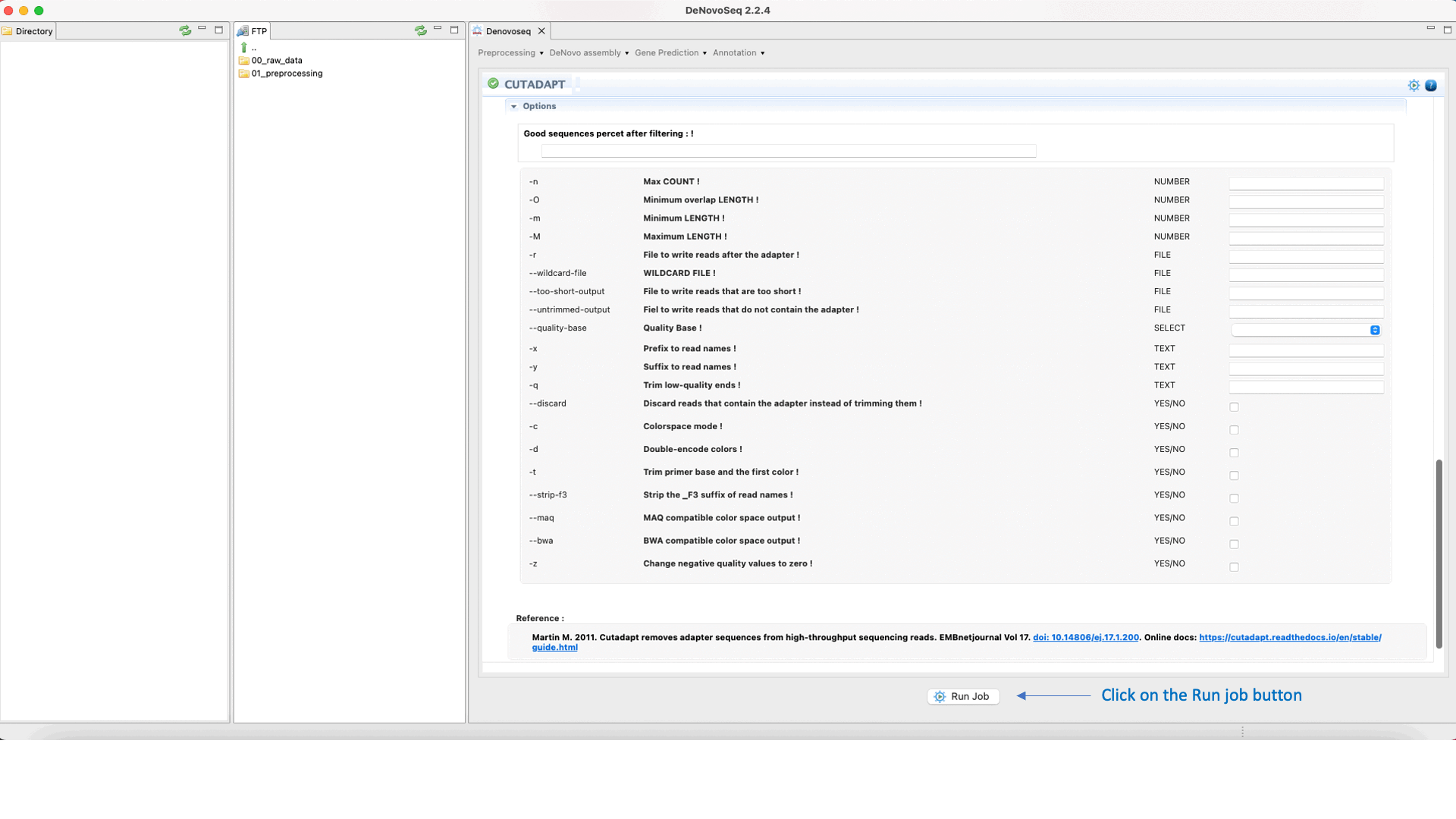
Figure 9: Using the GPRO interface for Cutadapt.
- Trimming & Cleaning: Prinseq
Prinseq (Schmieder and Edwards 2011) can filter, reformat, or trim your sequencing reads. For further information, see the Prinseq manual at http://prinseq.sourceforge.net.
To run Prinseq go to [Preprocessing → Trimming & Cleaning → Prinseq] and follow Fig. 10.
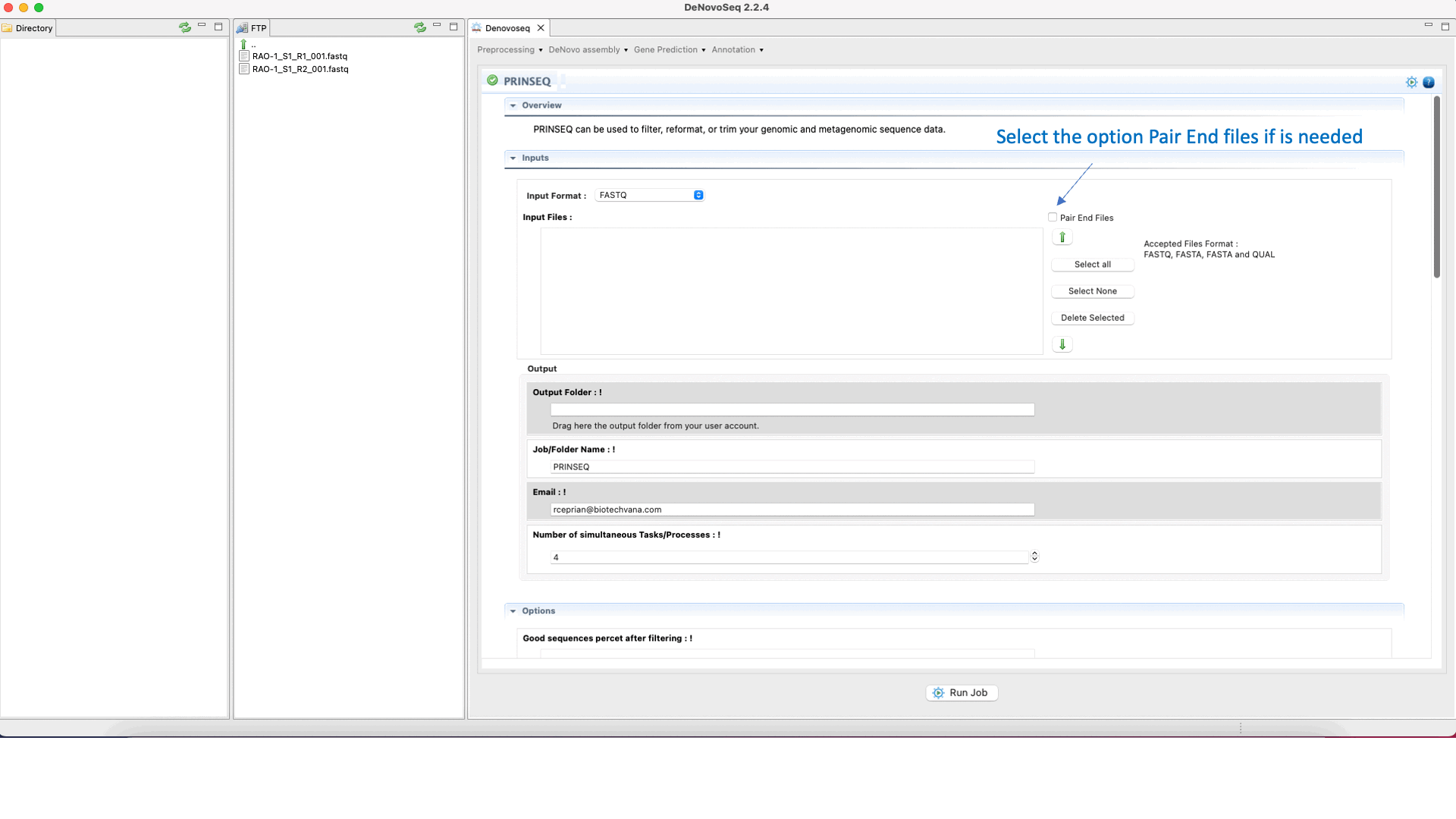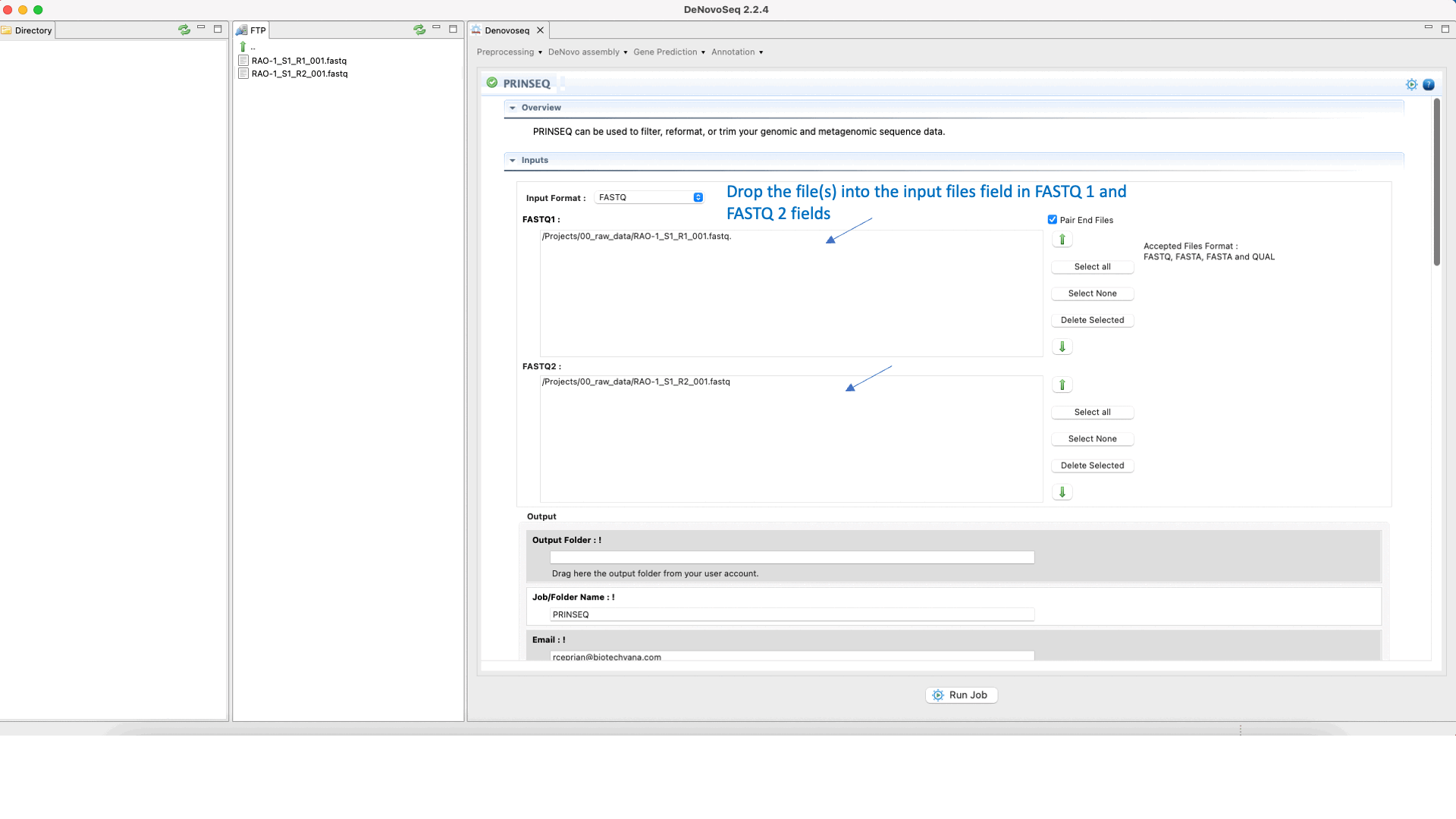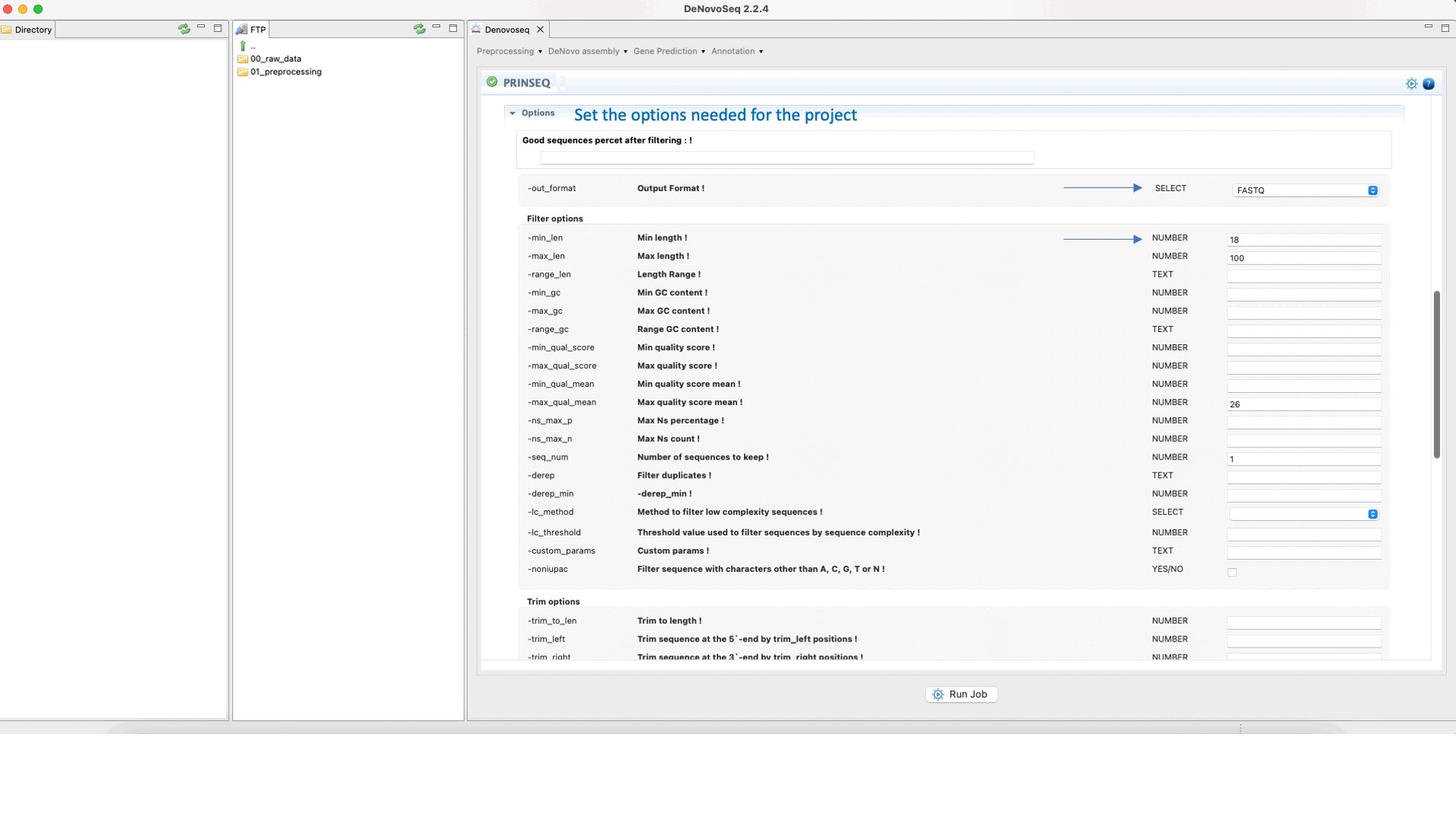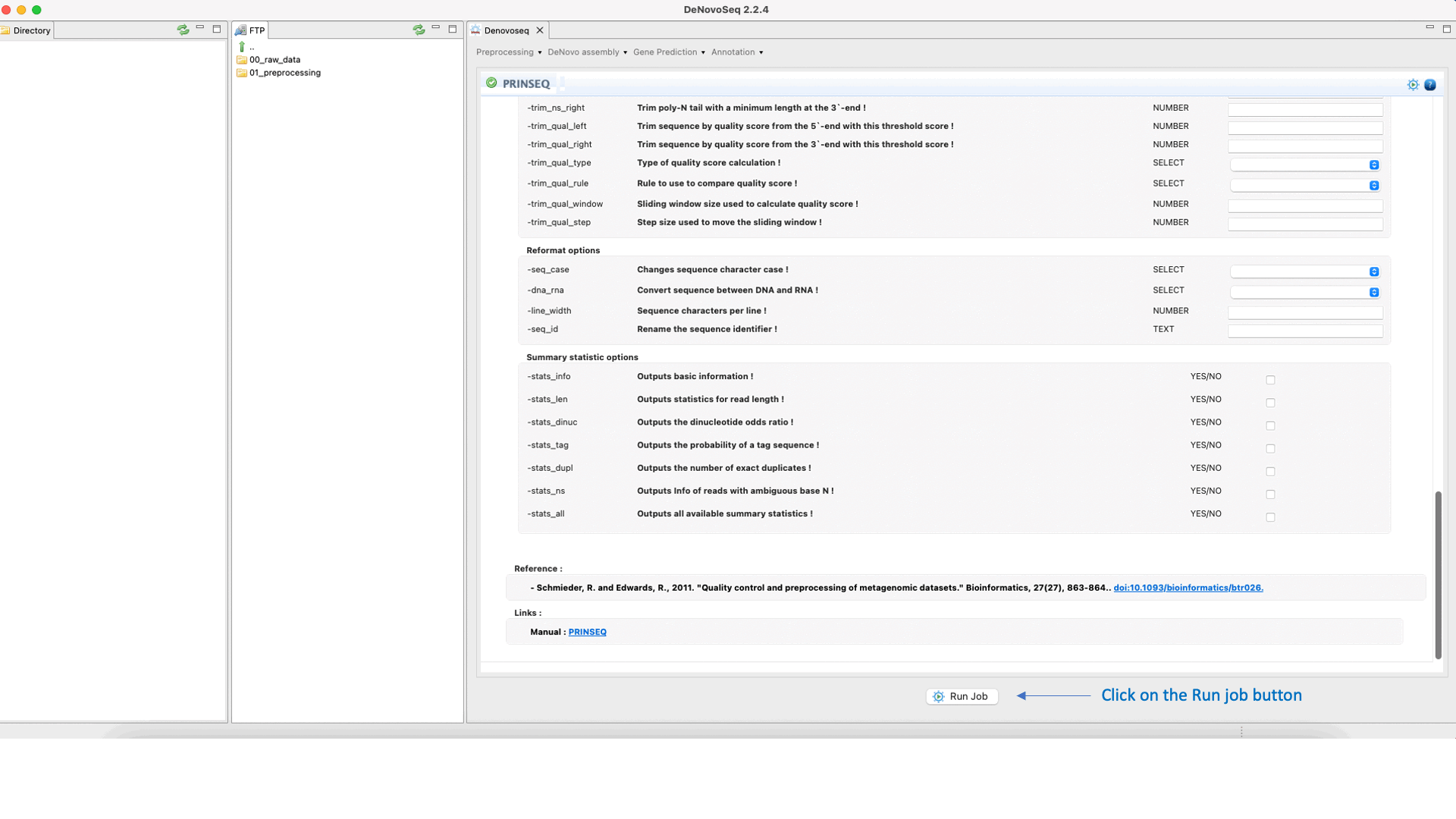
Figure 10: Using the GPRO interface for Prinseq.
- Trimming & Cleaning: Trimmomatic
Trimmomatic (Bolger et al. 2014) is trimming tool specific for paired-end and single-end reads obtained via Illumina’s NGS technology that can perform a variety of trimming tasks. For more information see the trimmomatic manual at "http://www.usadellab.org/cms/?page=trimmomatic".
To run Trimmomatic go to [“Preprocessing → Trimming & Cleaning → Trimmomatic”] and follow Fig. 11.
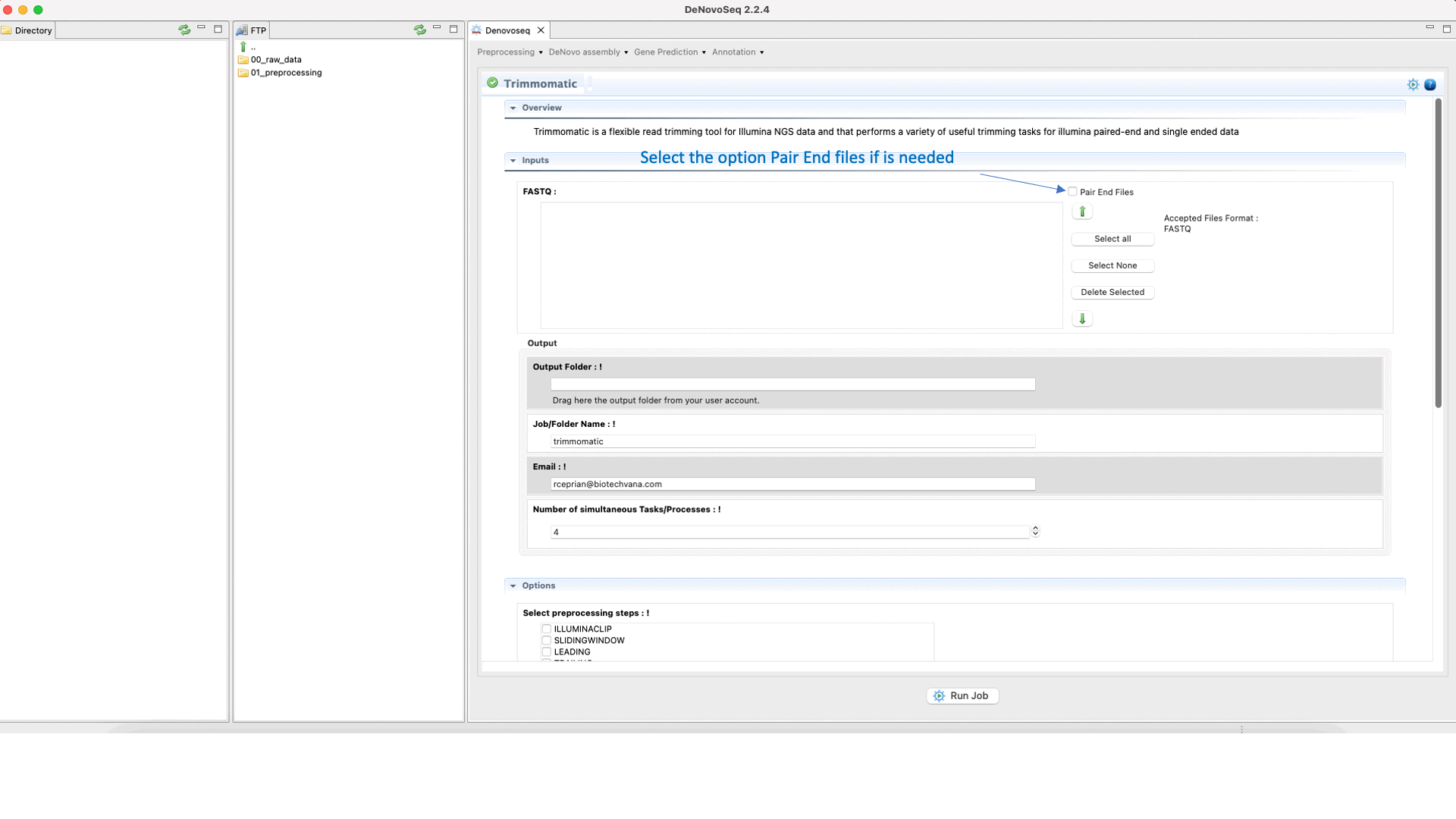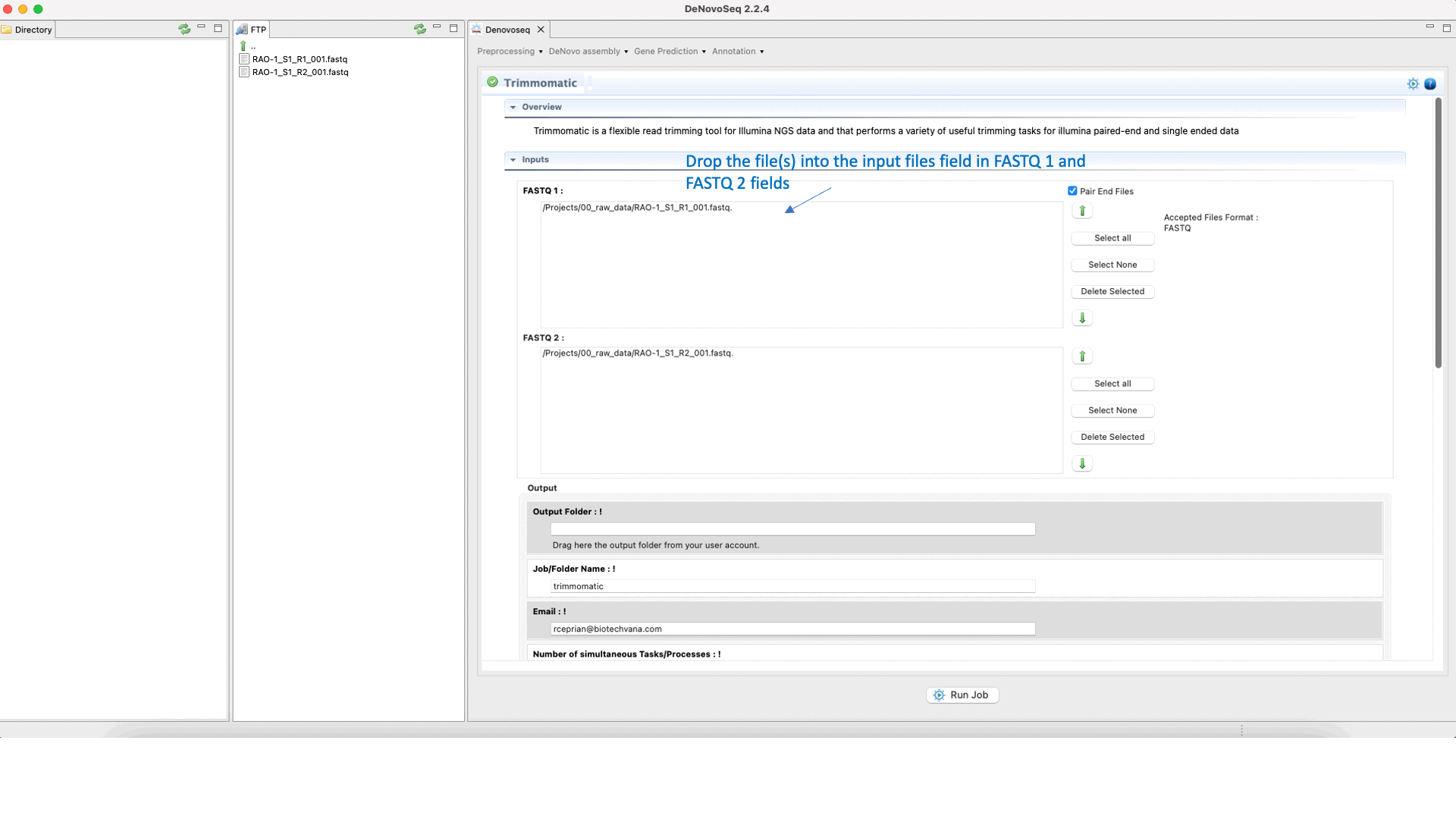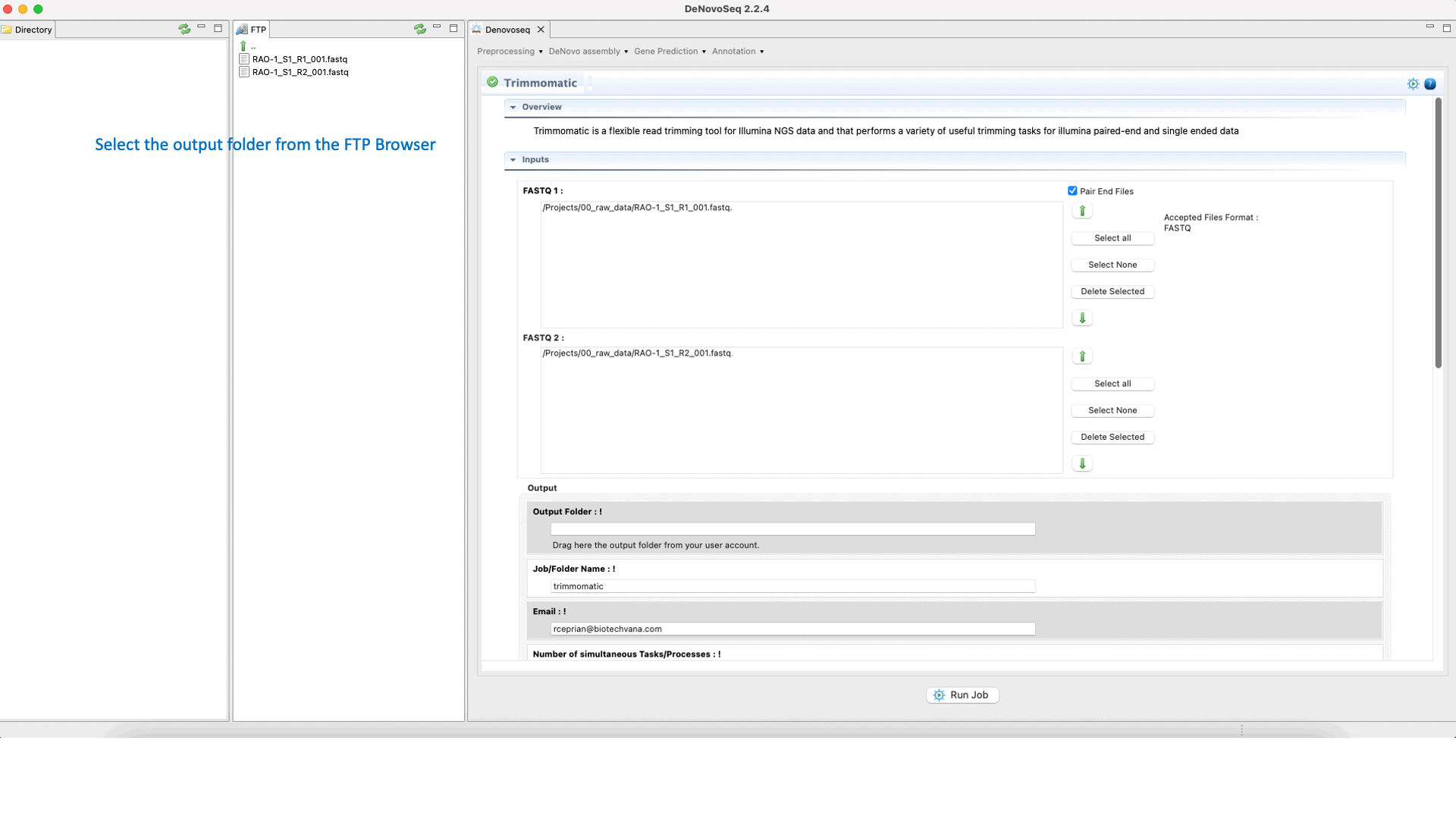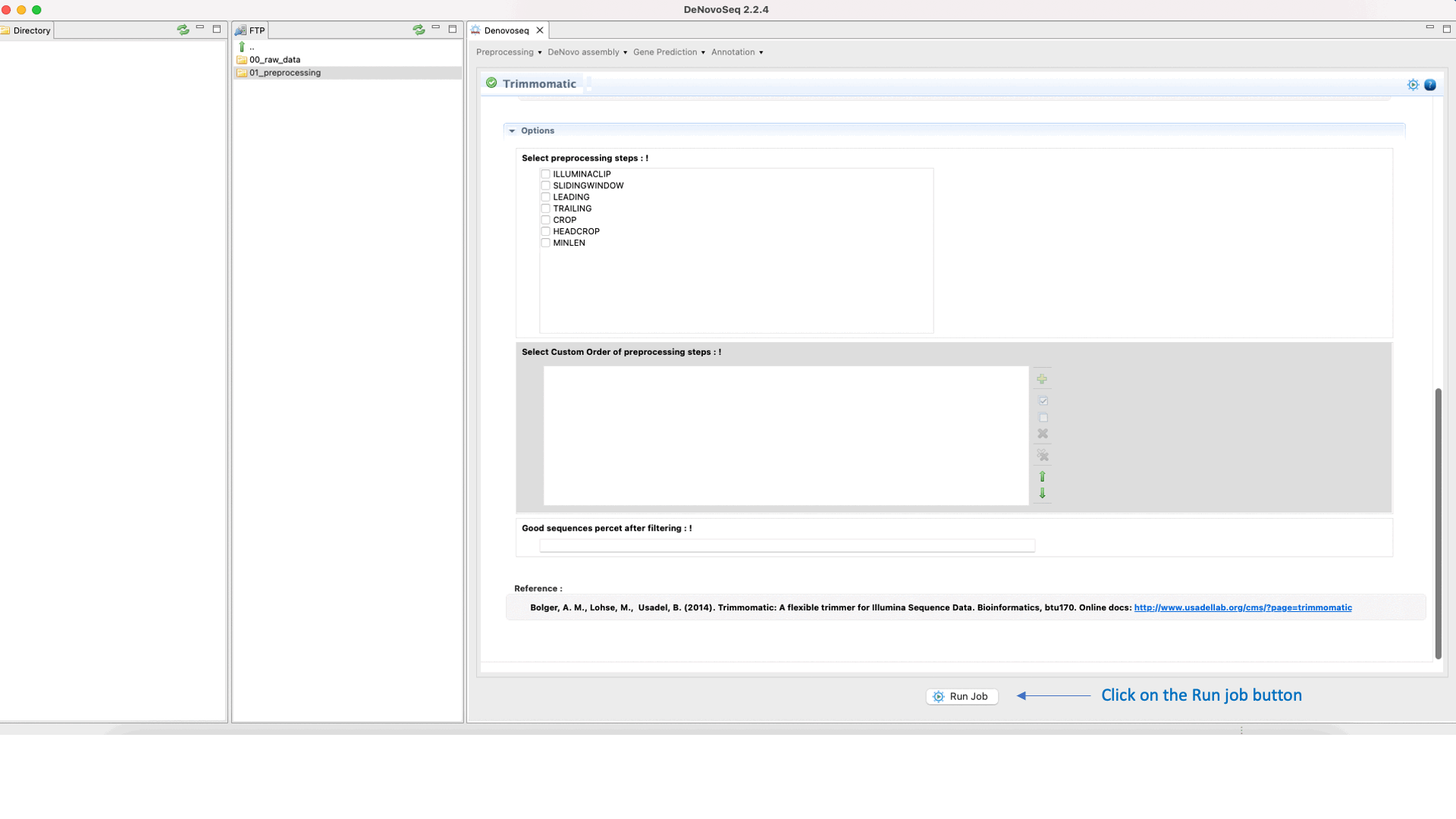
Figure 11: Using the GPRO interface for Trimmomatic.
- Trimming & Cleaning: FastxToolKit
FASTX-Toolkit (Hannon Lab 2016) iis a set preprocessing tools for Fasta/Fastq files:
To run any of the FastxToolKit go to [“Preprocessing → Trimming & Cleaning → Fastx-Toolkit”] and follow Fig. 12.
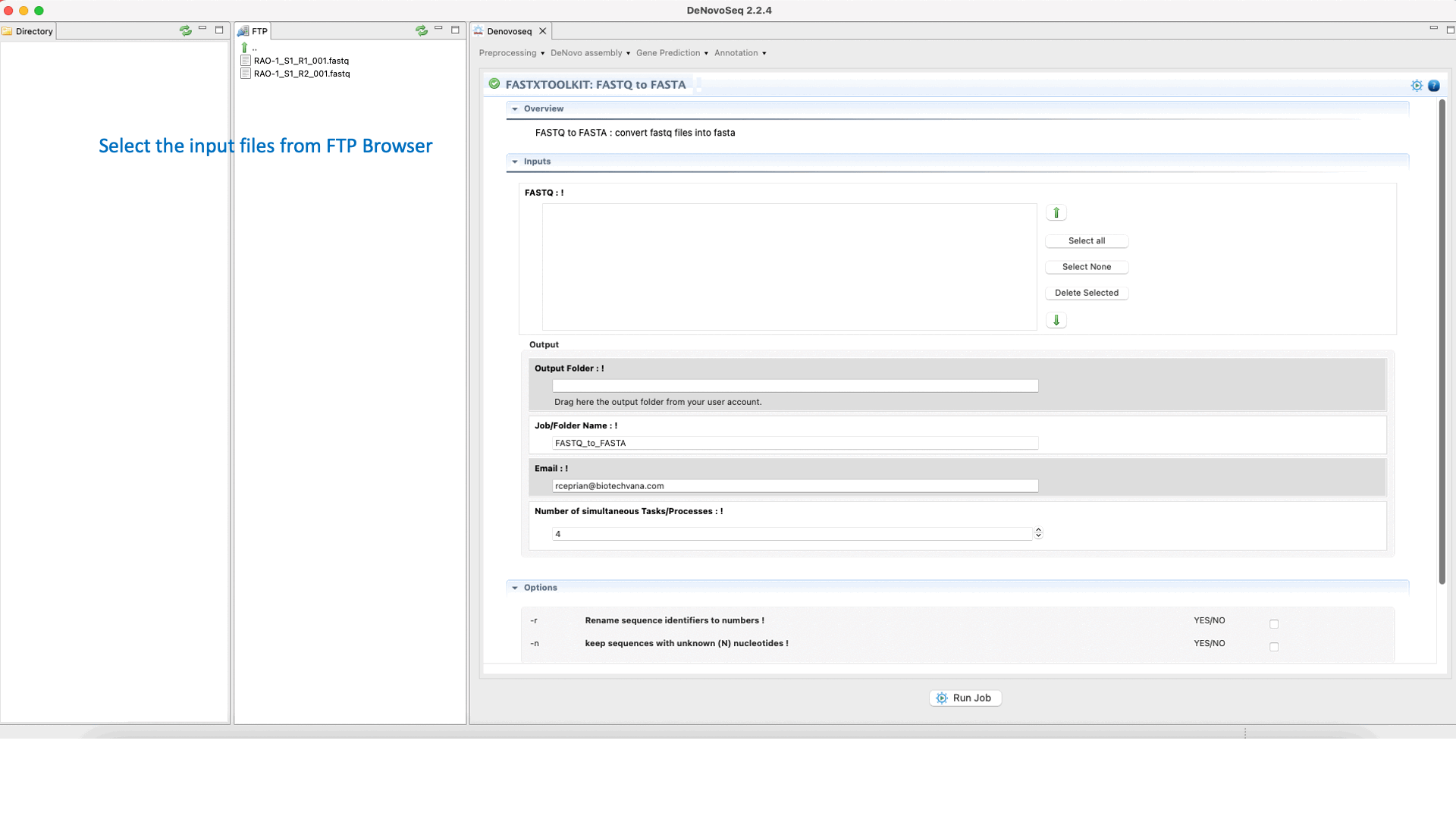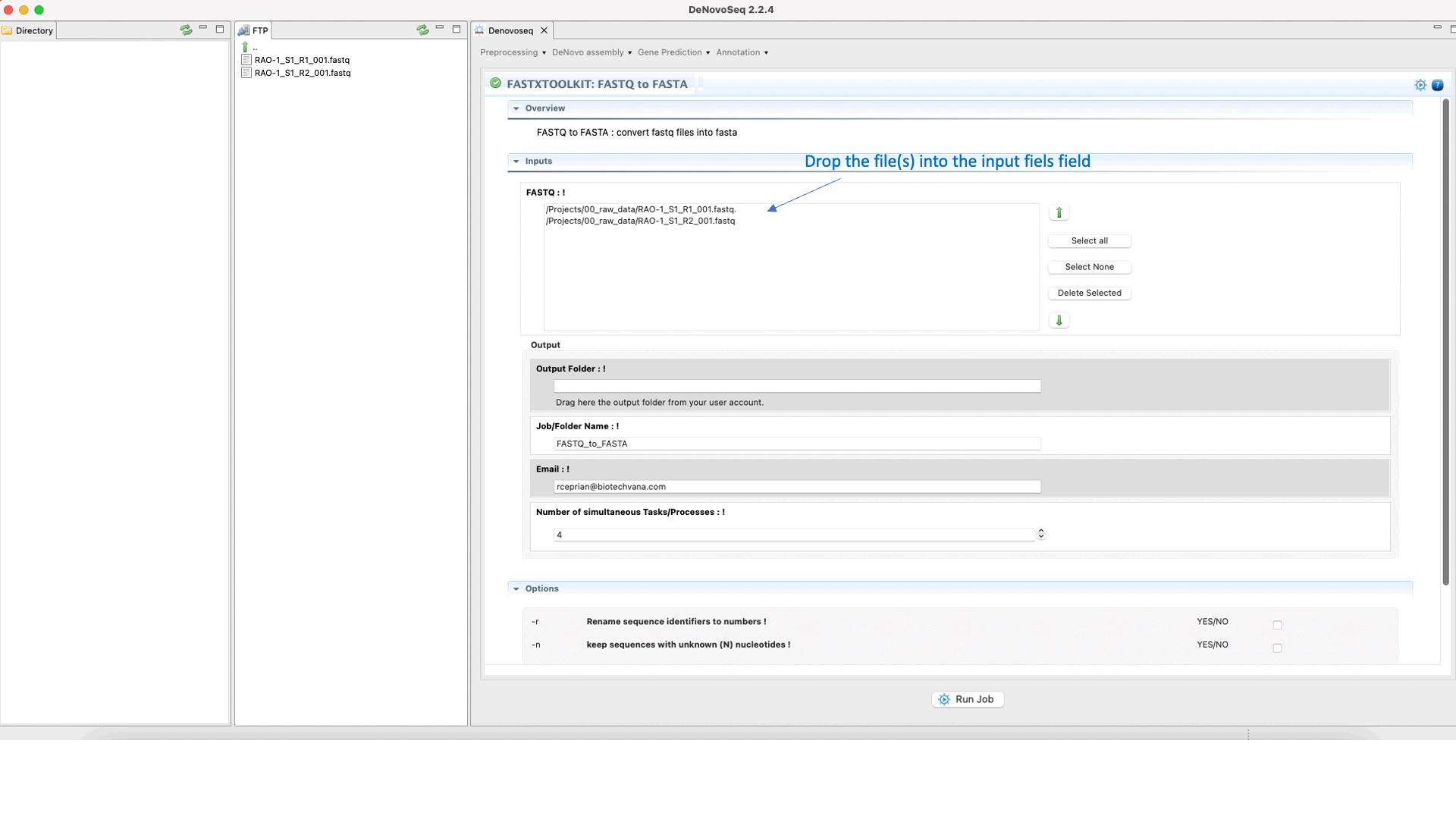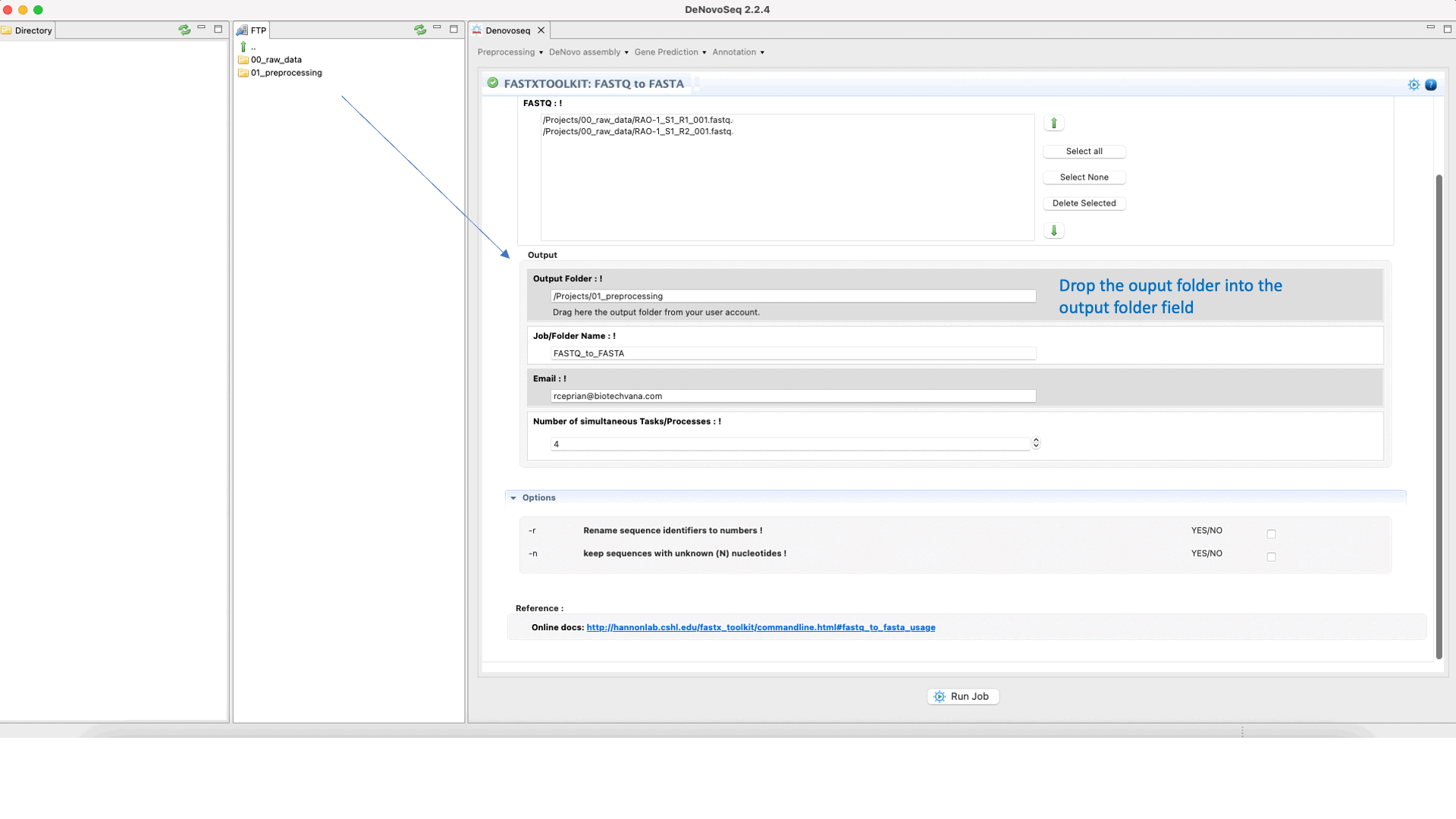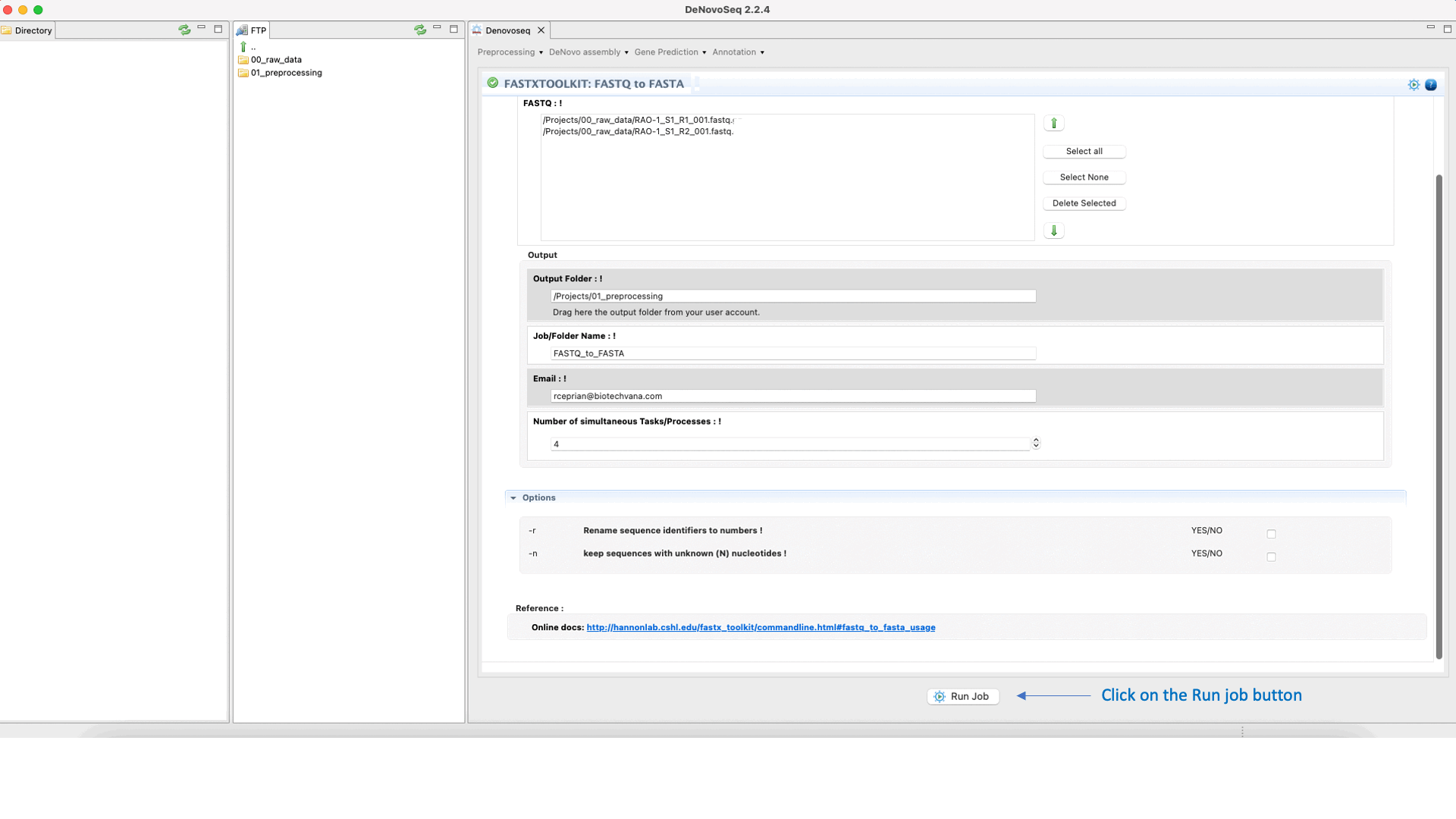
Figure 12: Example of GPRO interface for a tool (FASTXTOOLKIT: FASTQ to FASTA) of Fastx-Toolkit.
- PrepSeq: FastqCollapser
FastqCollapser is used to remove duplicate reads from fastq files based on their sequence content.
To run FastqCollapser go to[Preprocessing → PrepSeq → FastqCollapser] and proceed as shown in Fig. 13.
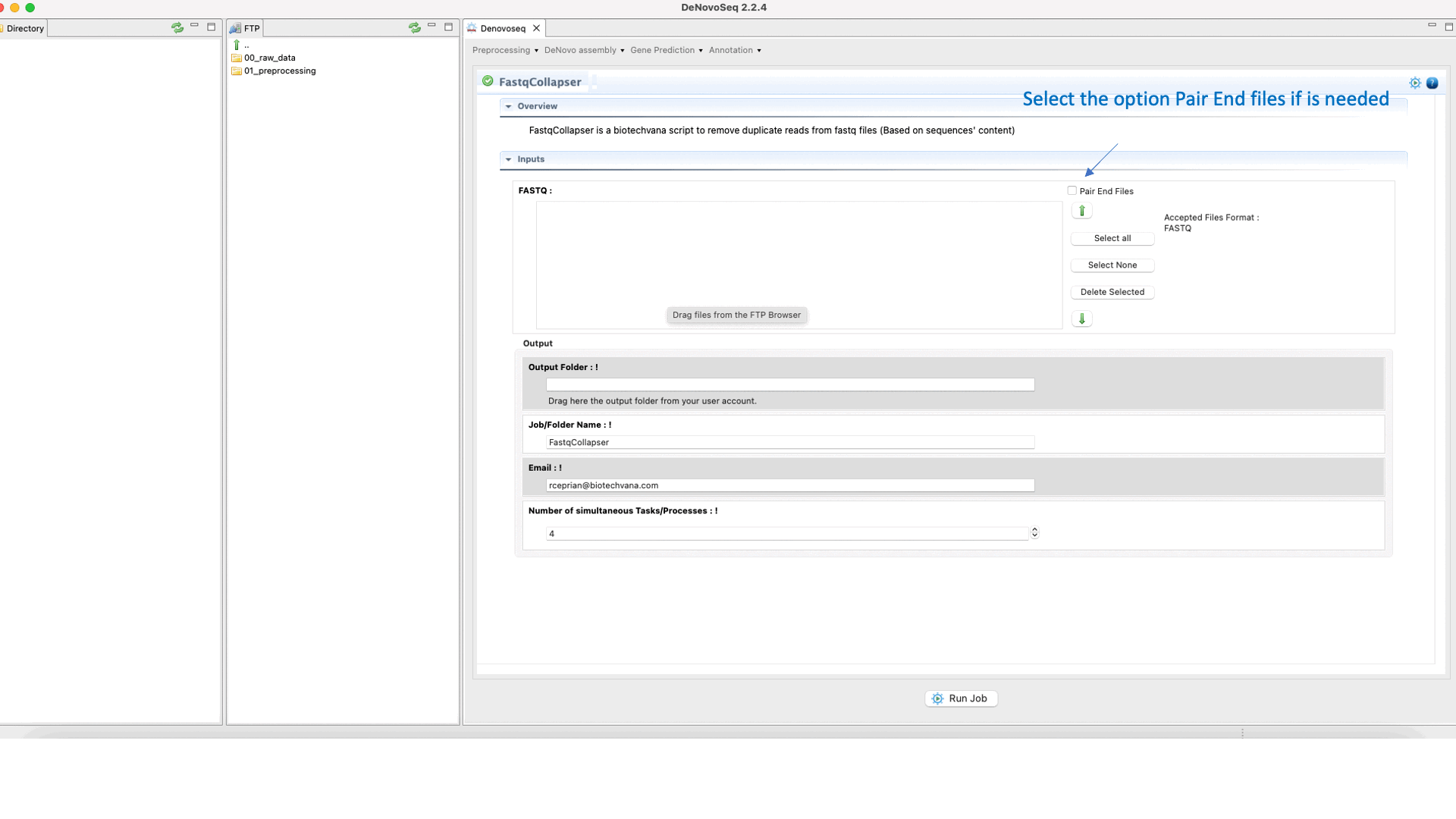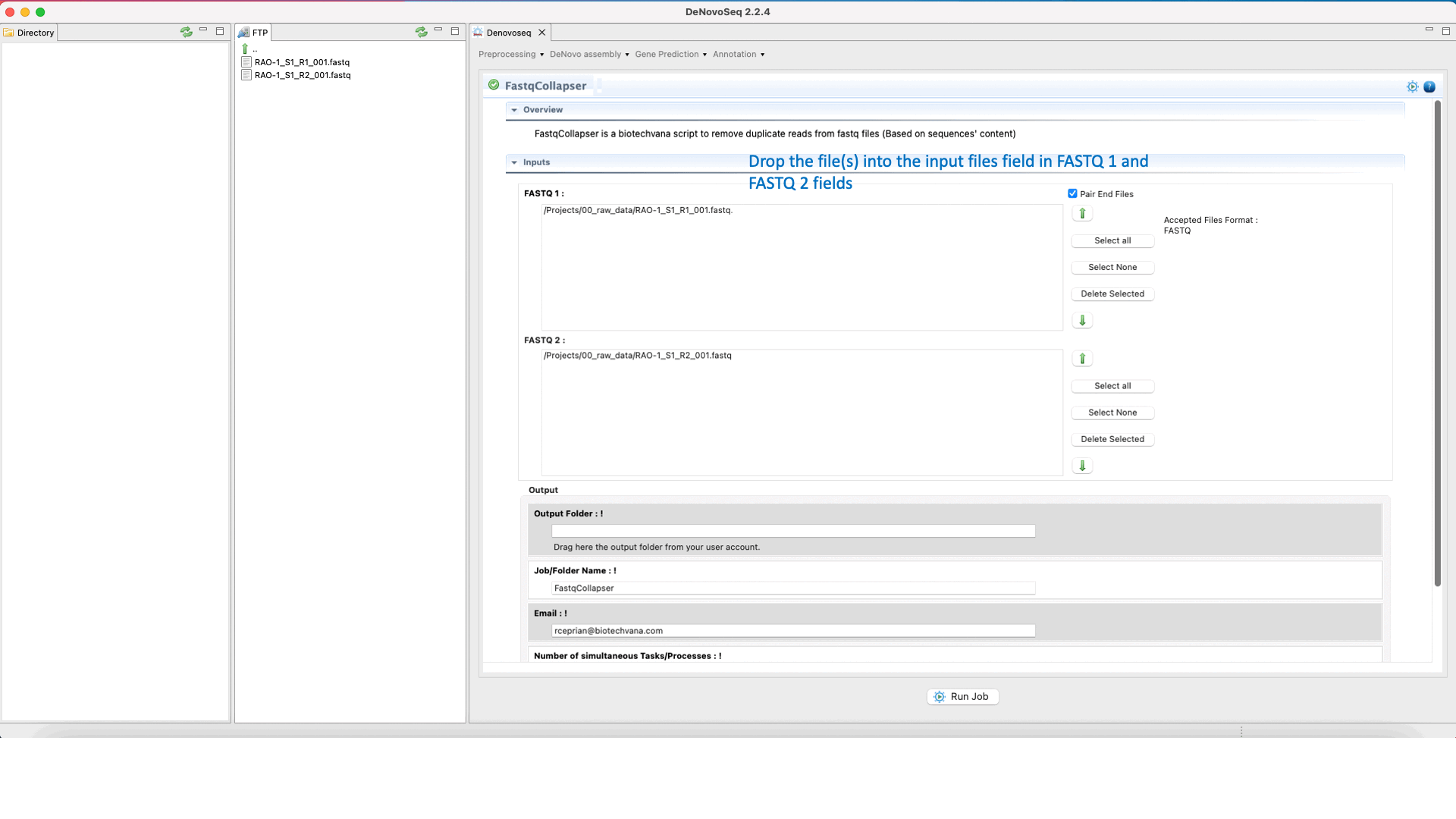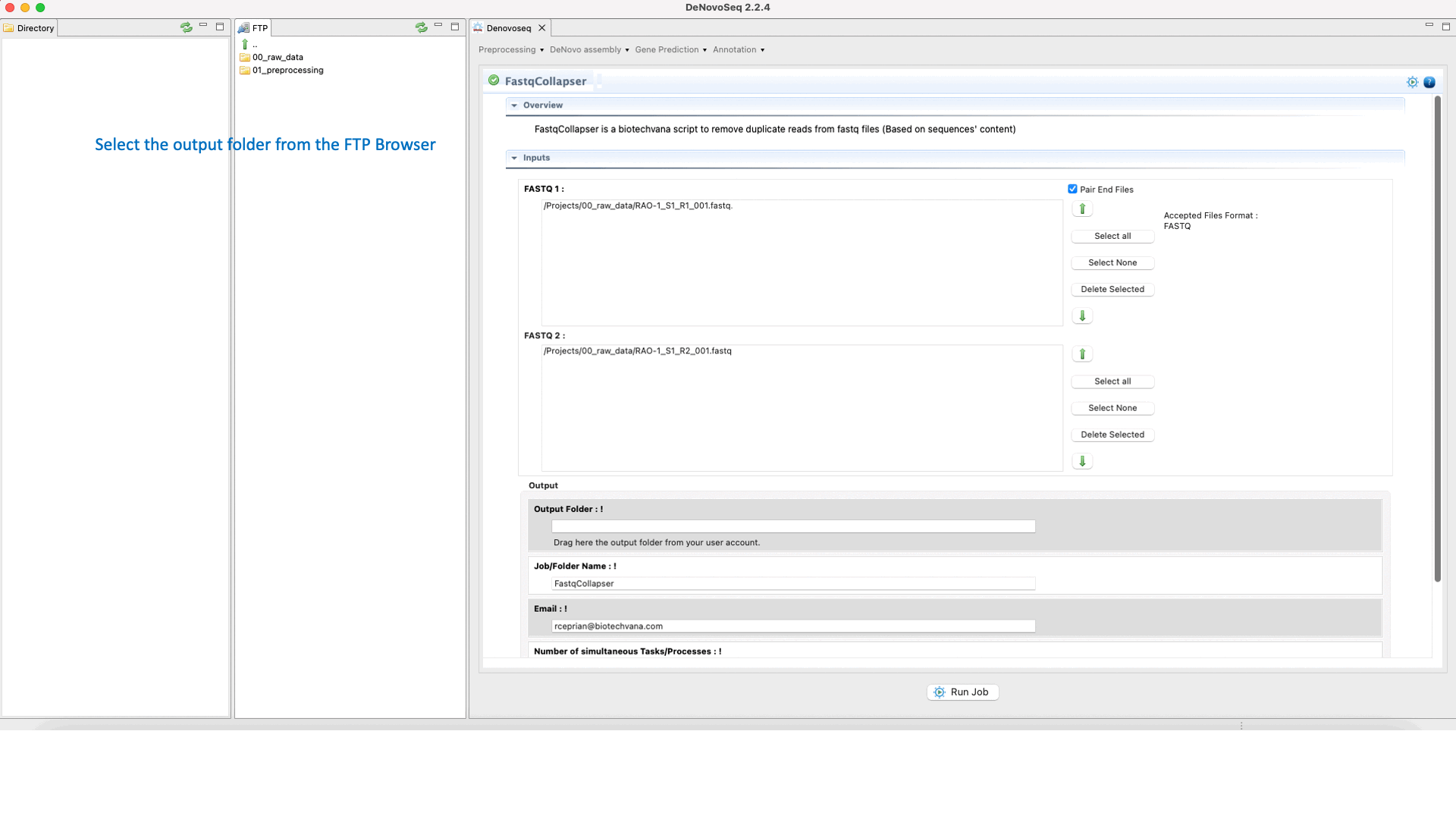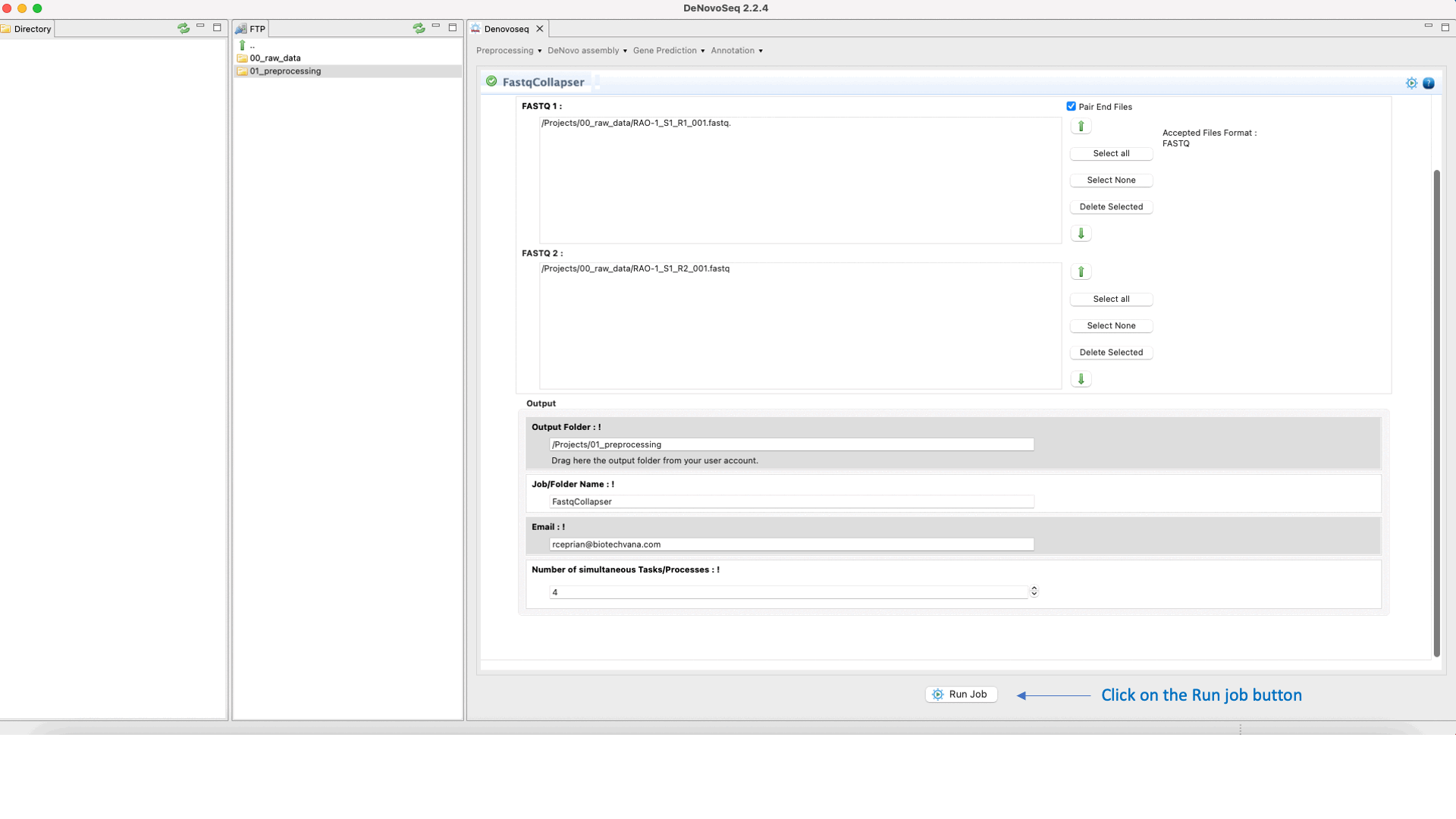
Figure 13: Using the GPRO interface for FastQCollapser.
- Trimming & Cleaning: FastqIntersect
FastqIntersect is a script that compares the information of two pair-end files that have been independently preprocessed and the information on both files to edit them keeping only those reads, and in the same order, that are present in both files (mate reads). This tool used when the number of reads obtained does not match the output of the execution of any preprocessing tool in each file individually the other. This is because assembly/mapping processes require that the files match in the number and the sort of reads. Please note that both Prinseq and Trimmomatic already have a function to intersect reads by ticking the ‘pair end files’ box. Thus, FastqIntersect will only need to be run in either those cases where the ’pair end files’ box has not been selected. FastqIntersect will also not need to be used when Cutadapt has been used, since this tool does not implement intersecting functions.
To run FastqIntersect go to [Preprocessing → PrepSeq → FastqIntersect] and follow Fig. 14.
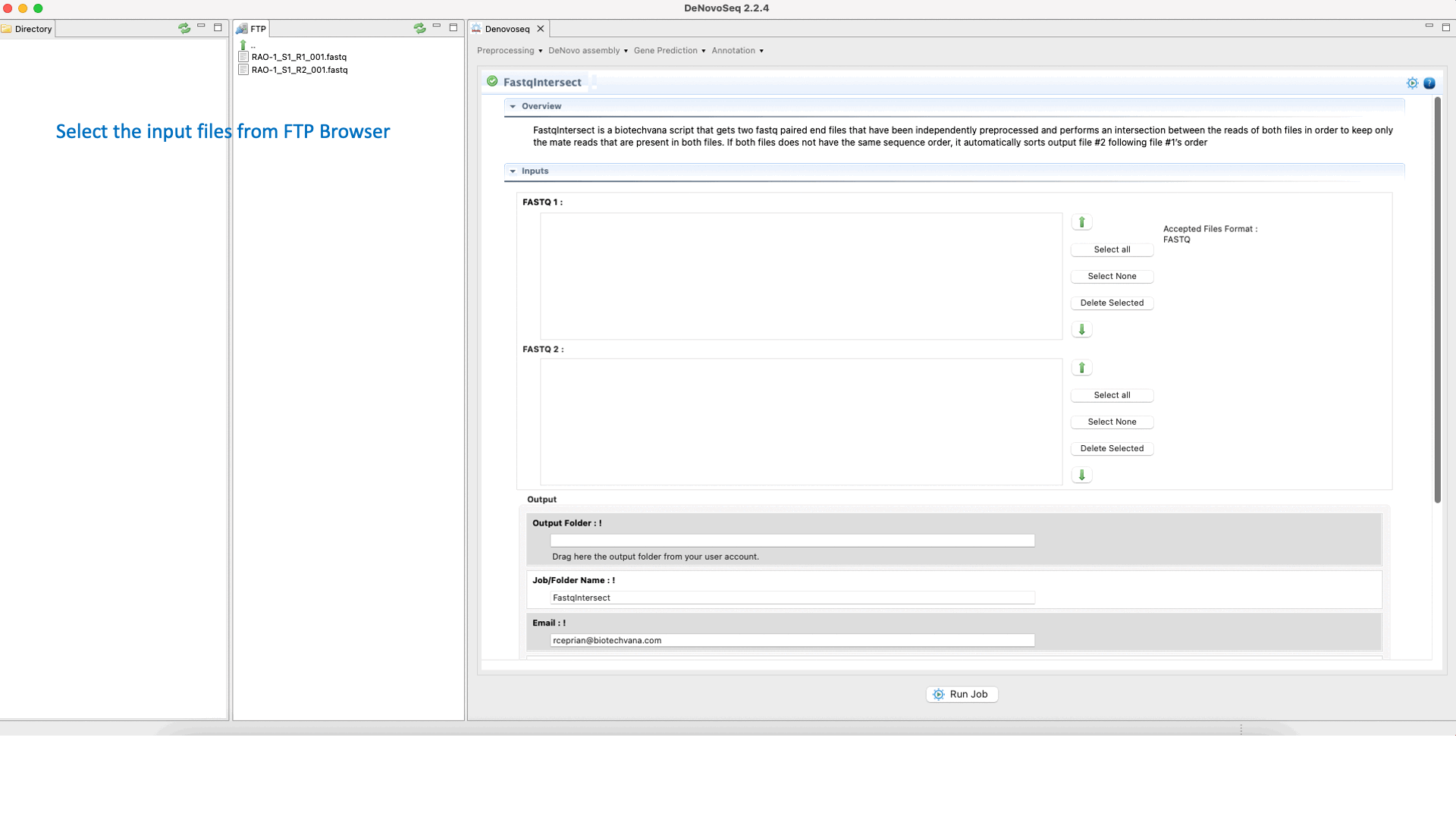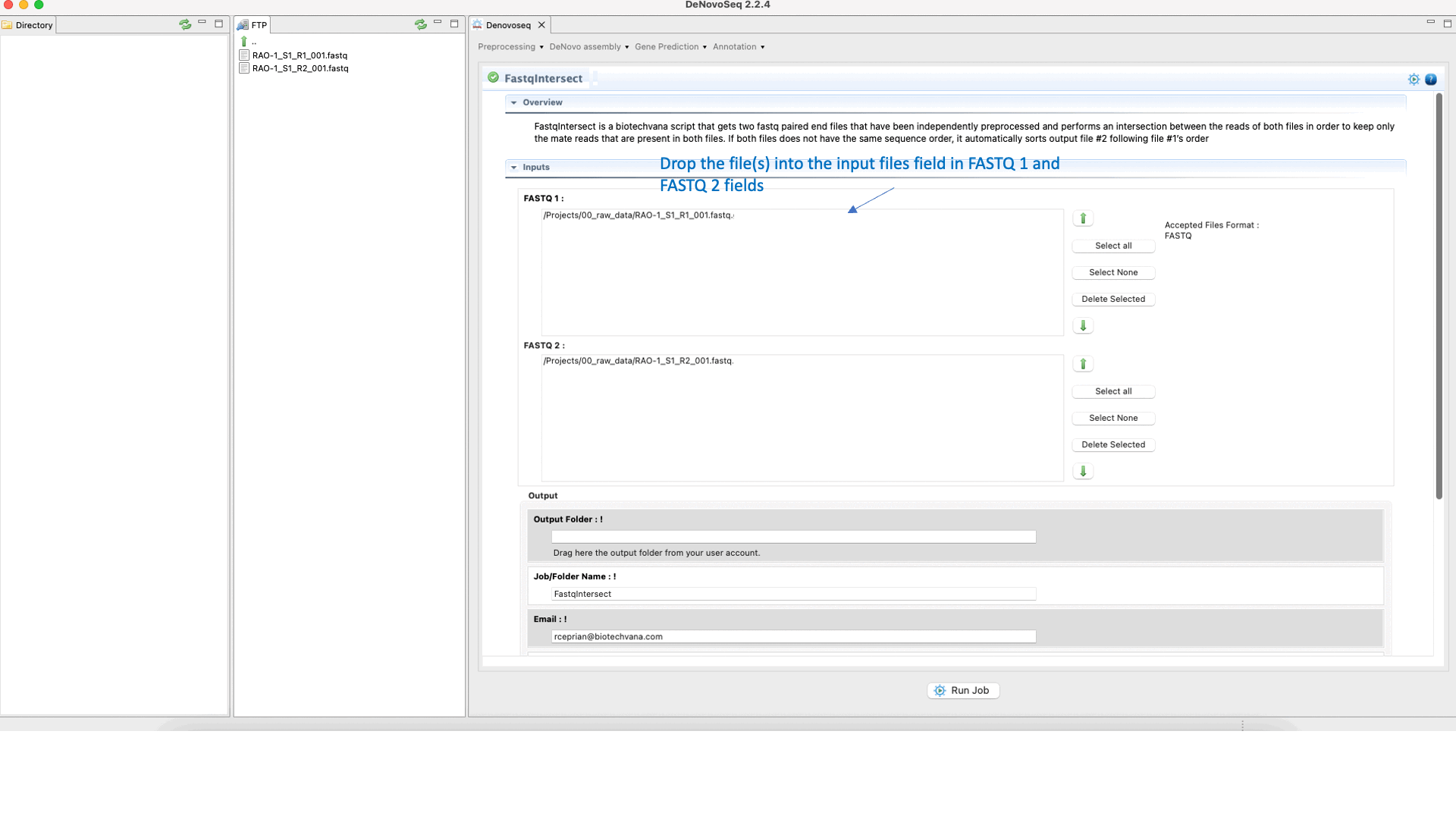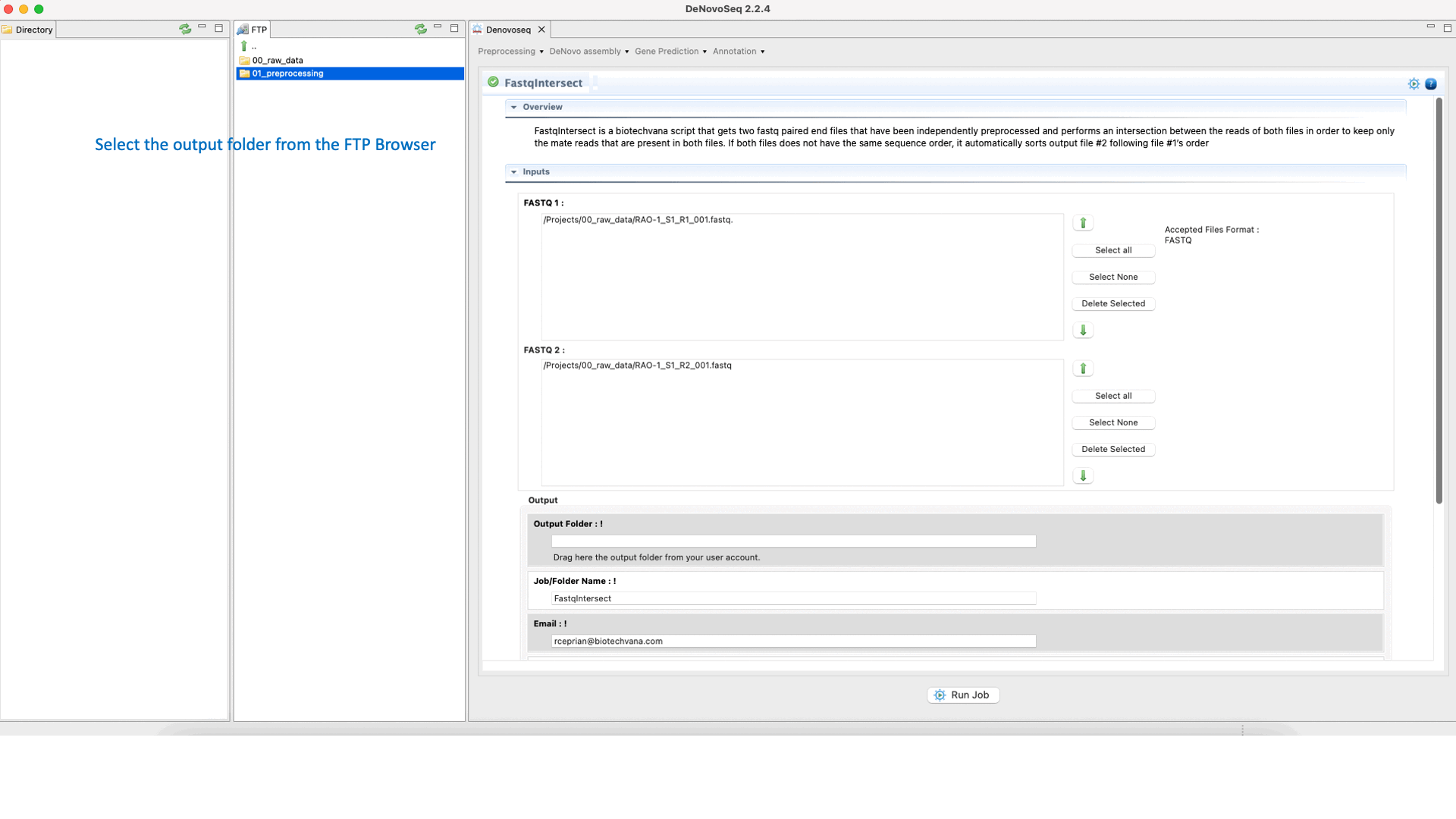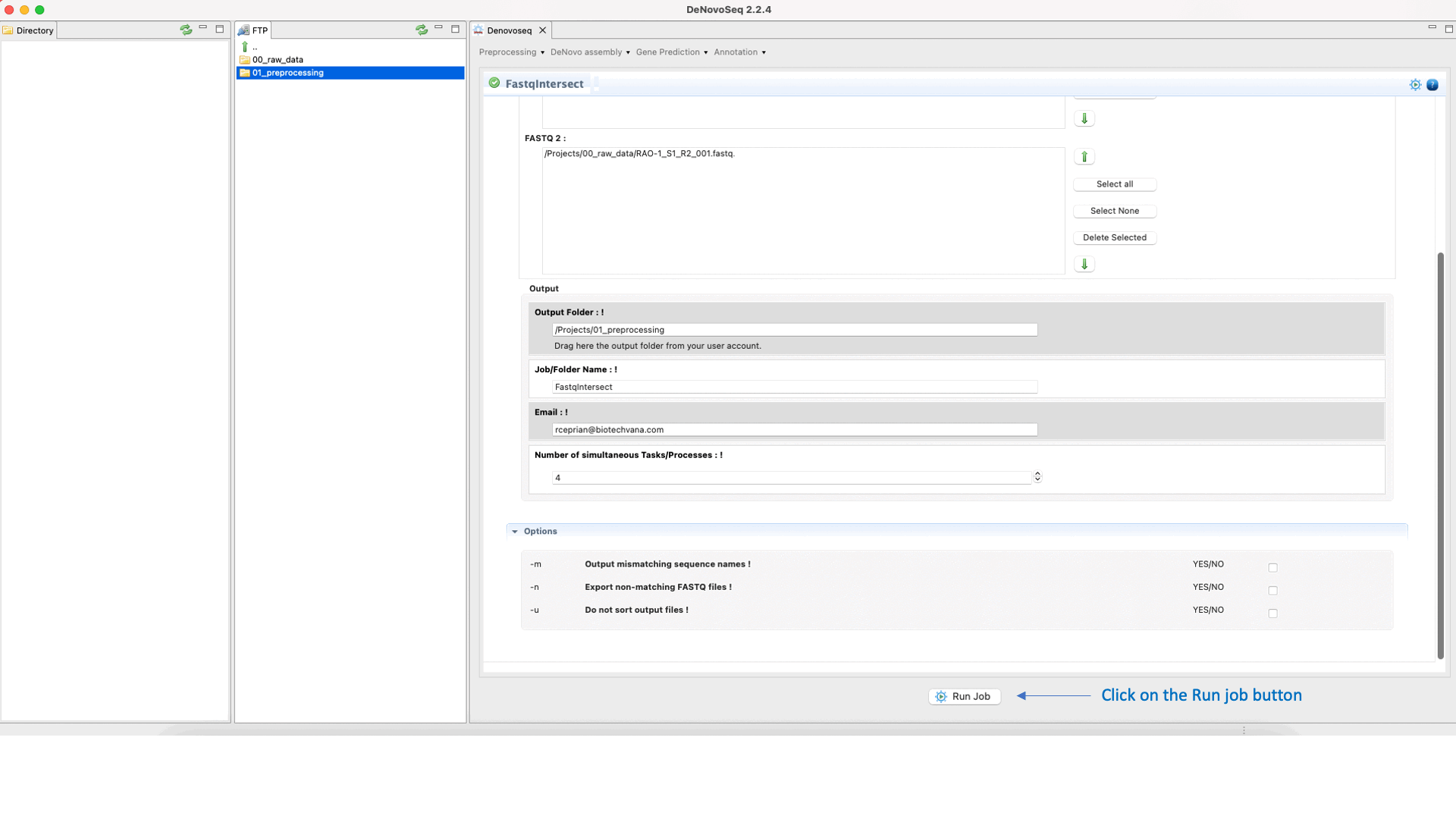
Figure 14: Using the GPRO interface Fastqintersect.
DeNovoSeq requires an An input configuration file to create a noew assembly project and guide the assembly process. To this end, users need to go this path [DeNovo Protocols → Input configuration file] for accessing an interface where the user either can download a previously existent configuration file with the experimental settings or may create a new one (indicating number and type of fastq libraries, libraries, type of sequencing, insert size, etc) as shown in Figure 15.
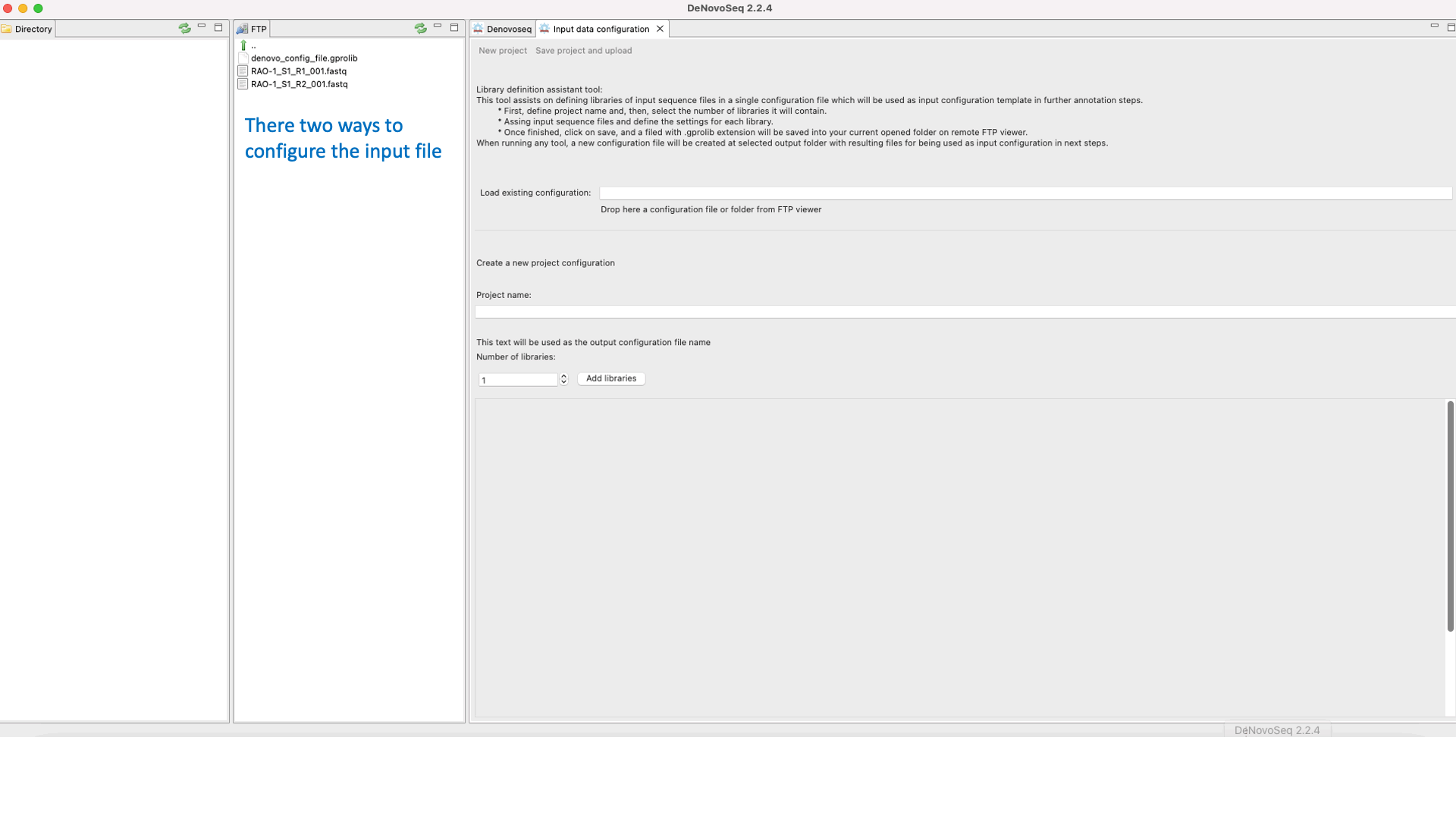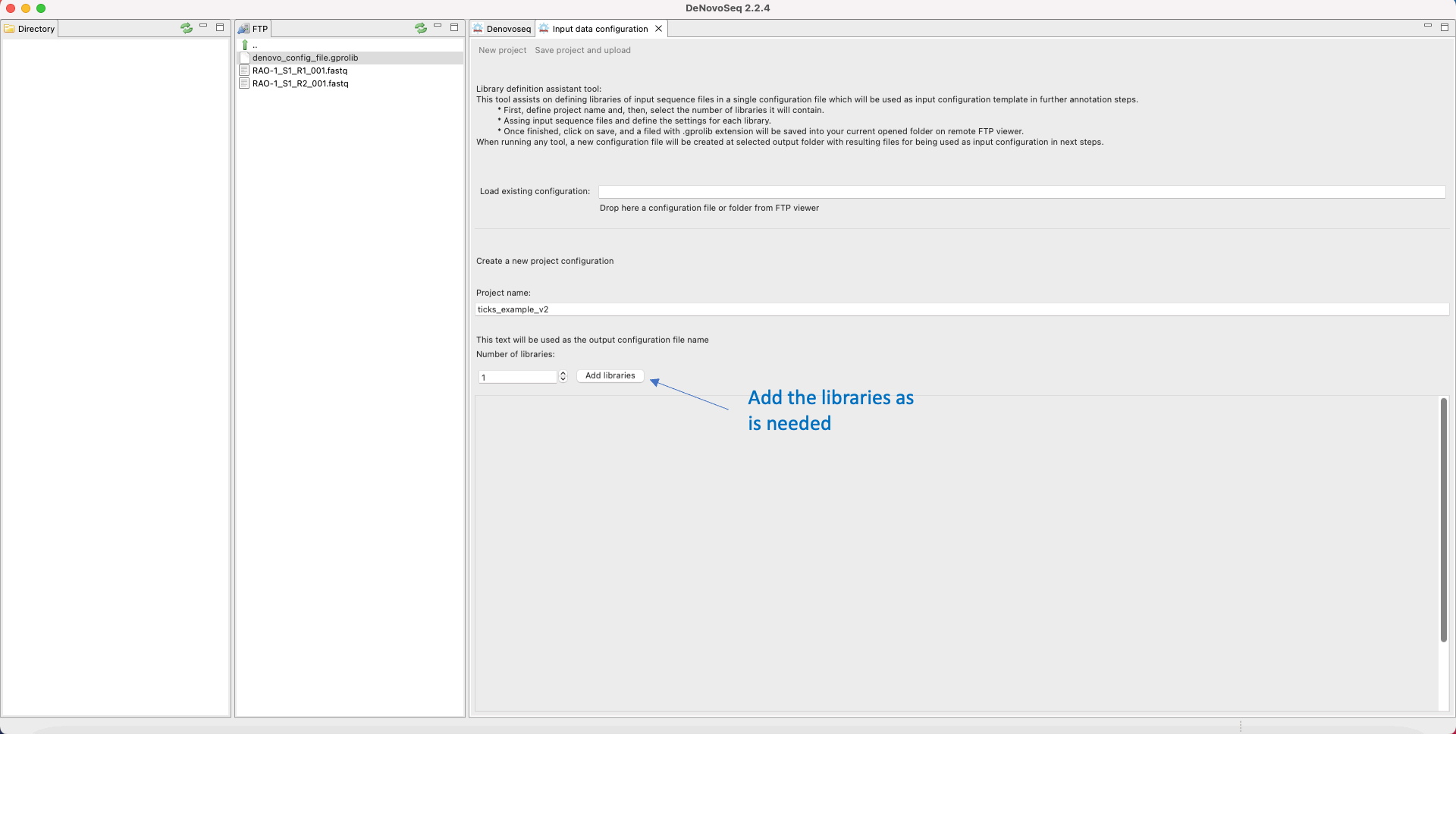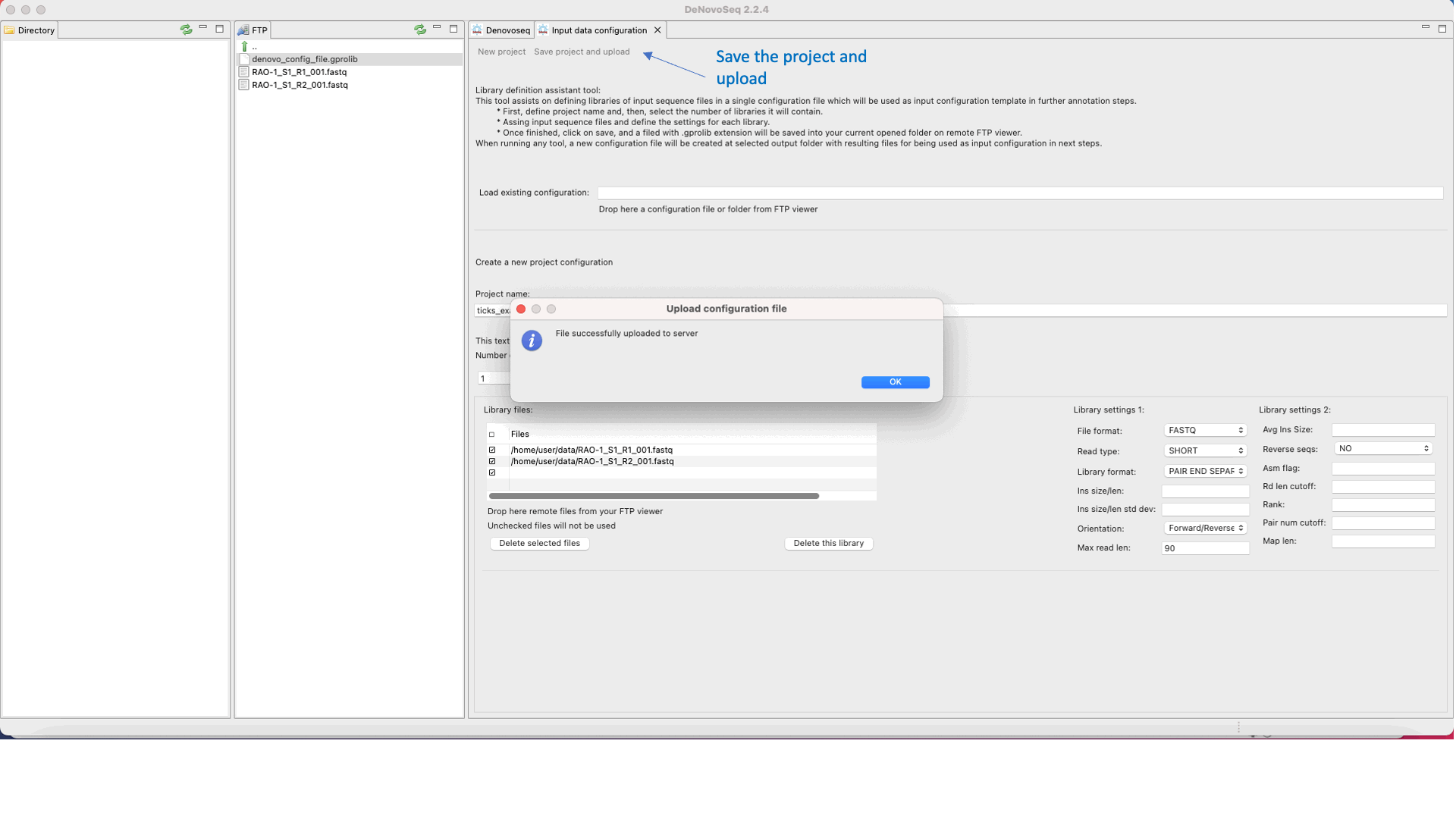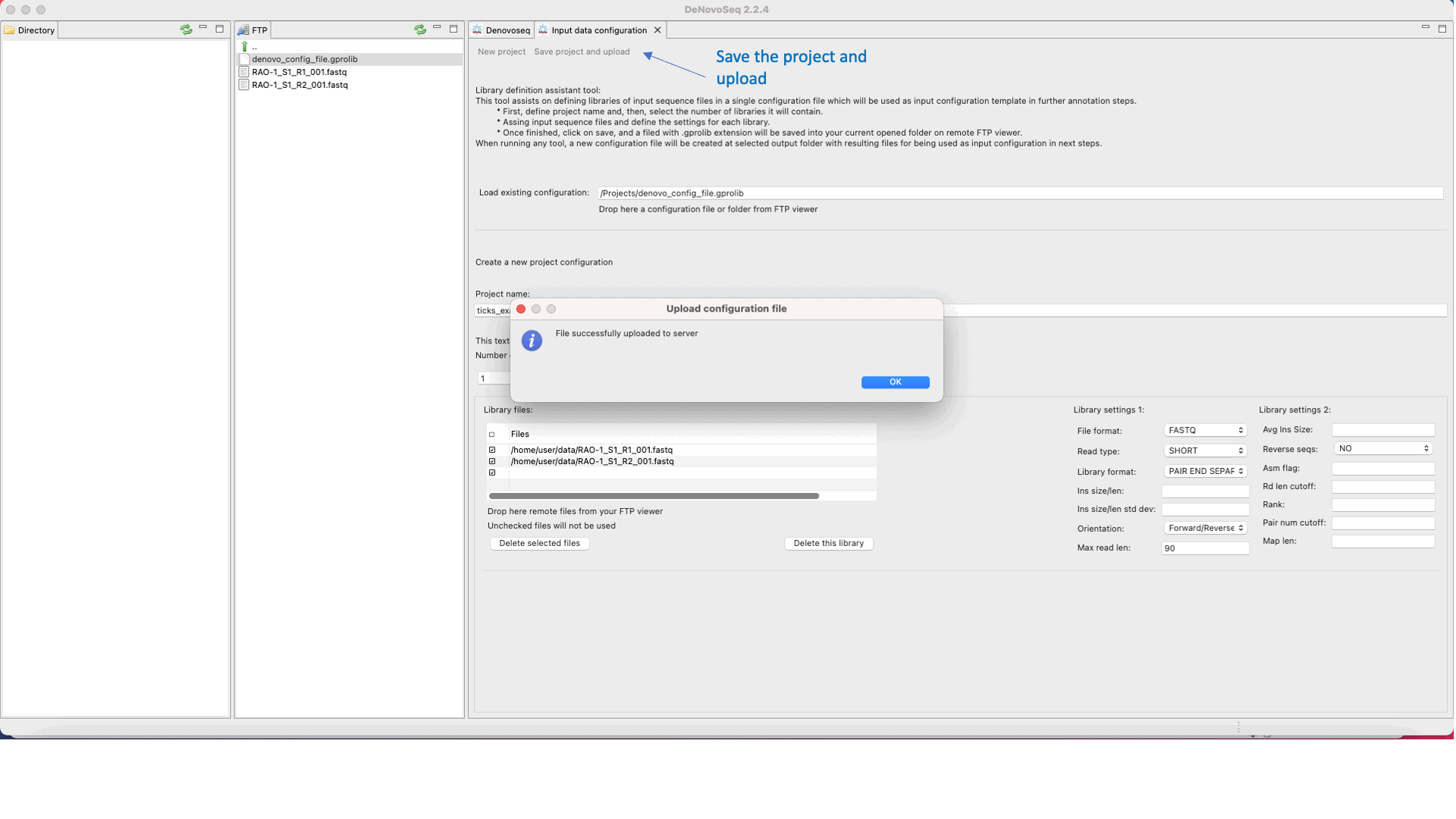
Figure 15: DeNovoSeq interface for configuring the input file needed by assemblers..
DeNovoSeq implements interface solutions for six debruijn graph Assemblers; two of them (assemble transcriptomes and the four others focus on the genomes assembly
- Transcriptomes: Oases
Oases (Schulz et al., 2012) is a de novo transcriptome assembler powered by a Velvet assembler core to resolve transcripts from short read and long sequencing reads in the absence of any genomic reference. For more information, see the Oases manual at "https://www.ebi.ac.uk/~zerbino/oases/OasesManual.pdf"
To run Oases go to [De novo assembly → Assembly → Transcriptomes → Oases]and follow Fig. 16.
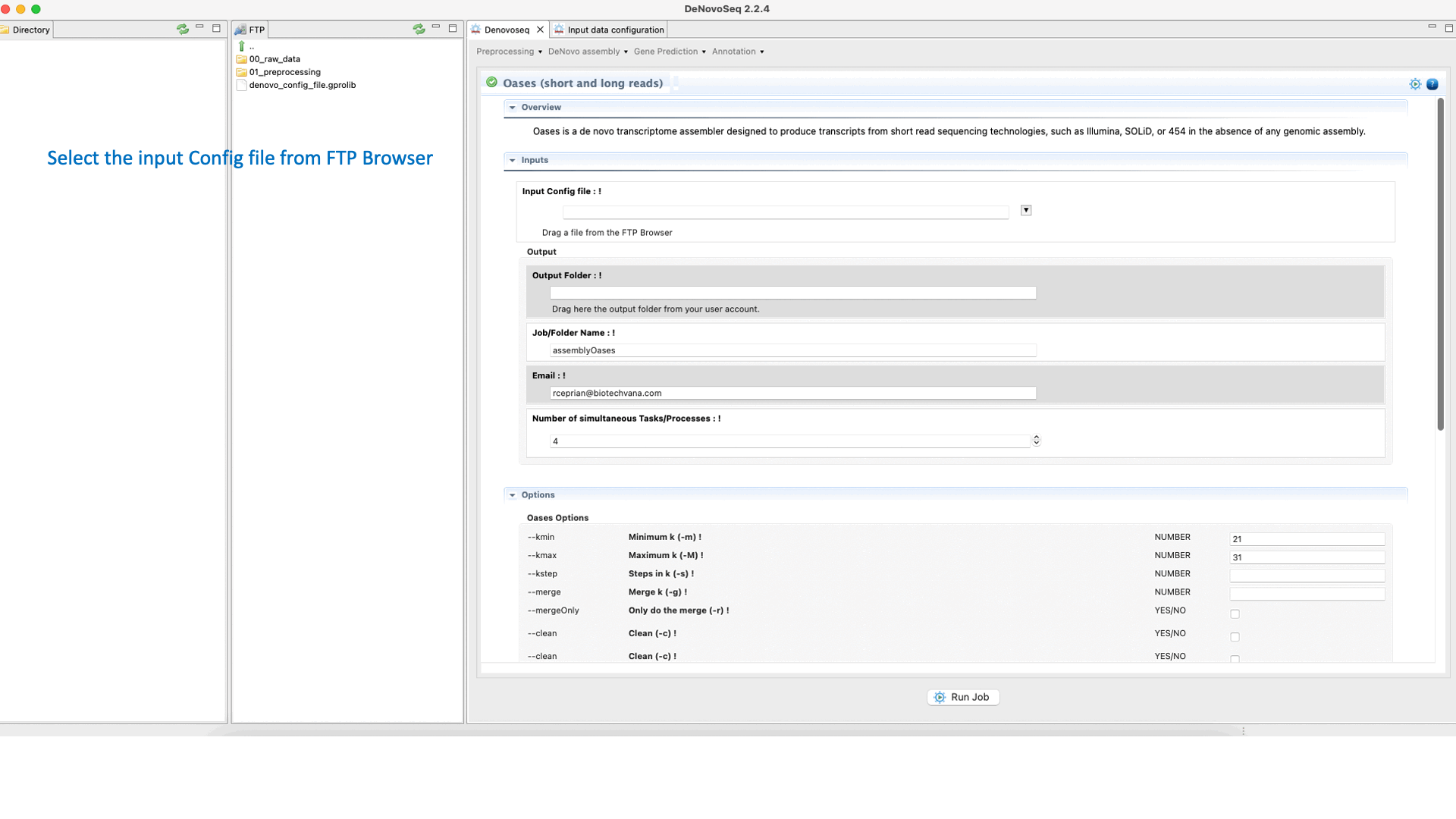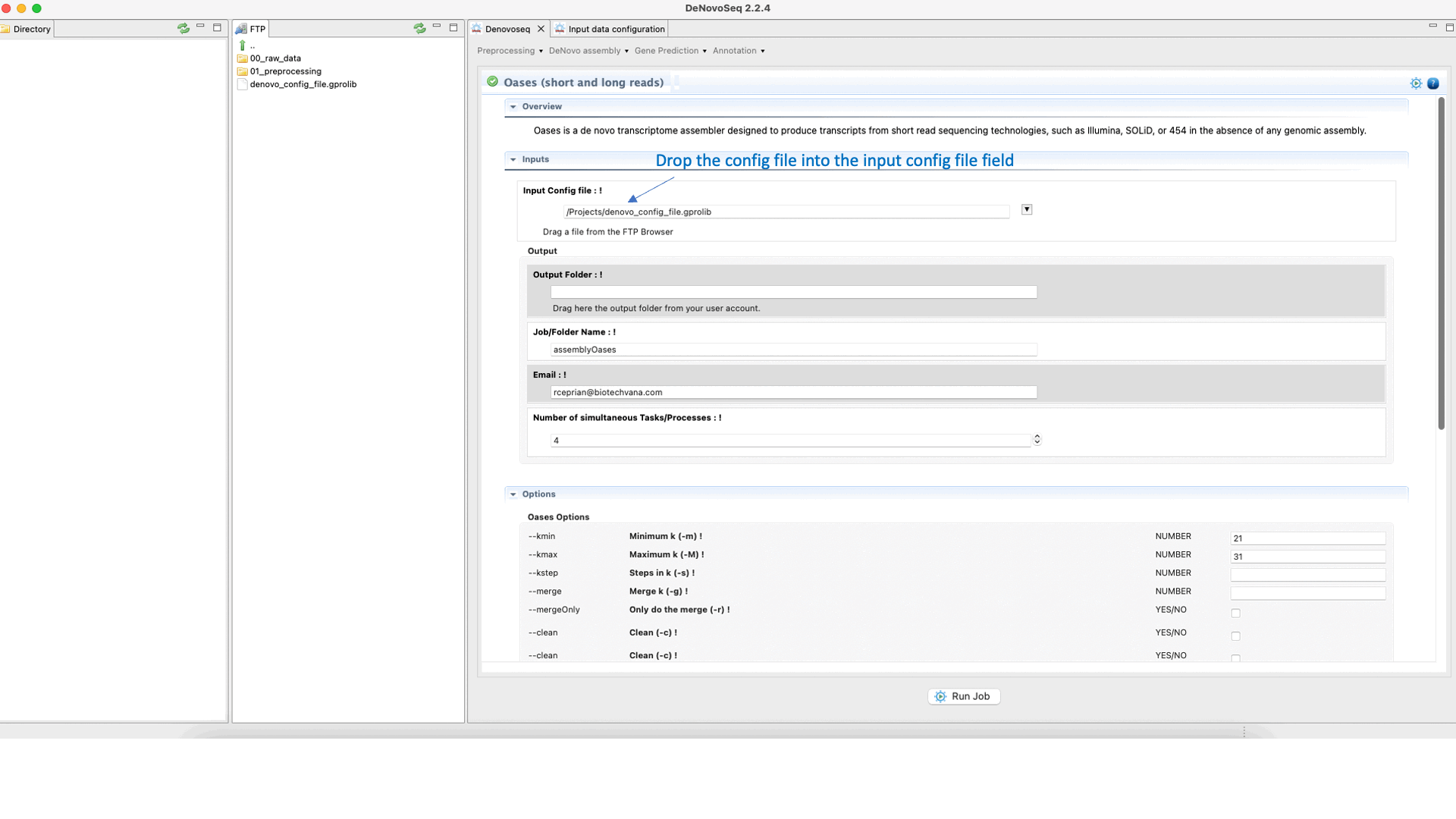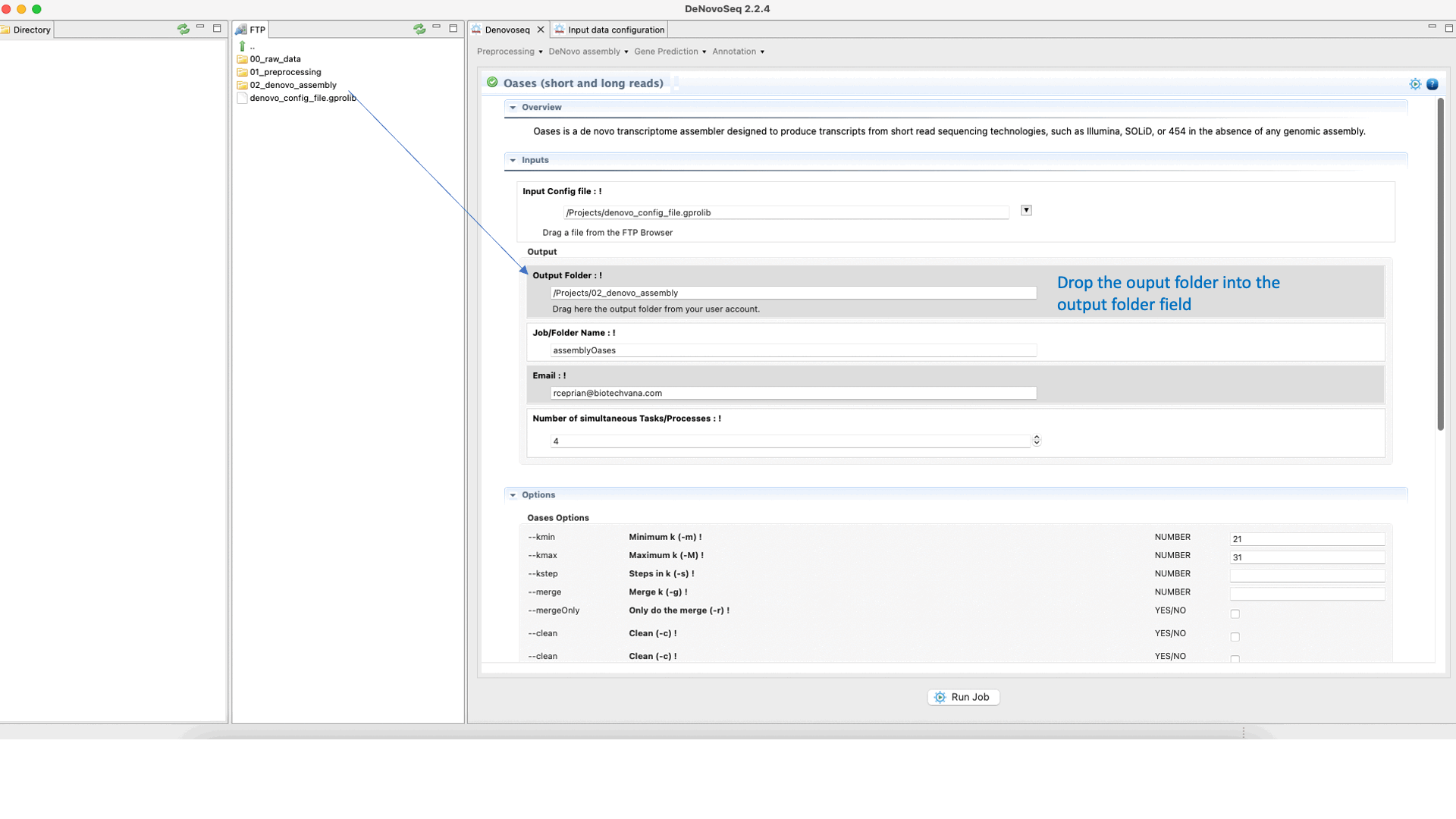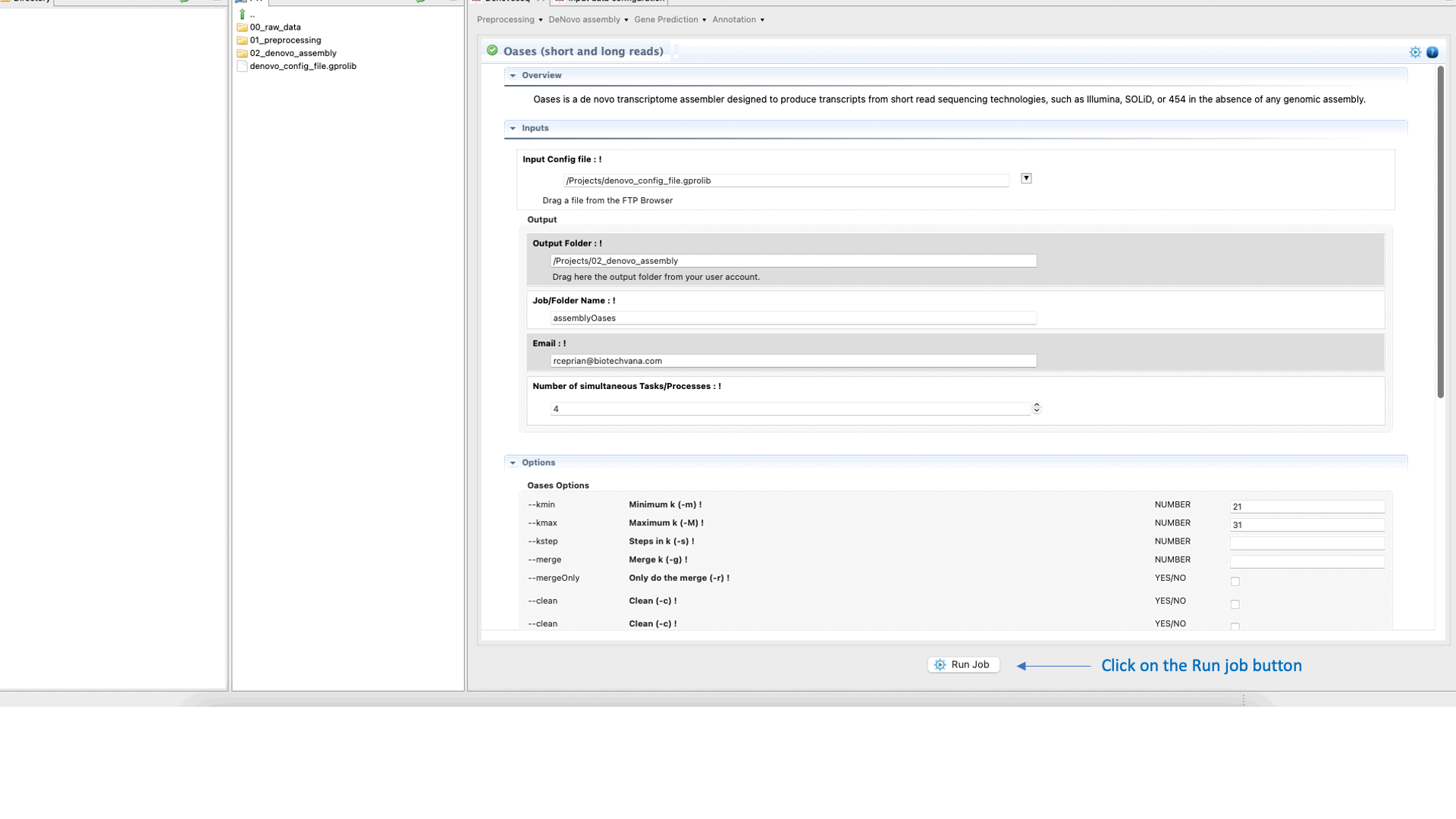
Figure 16: DeNovoSeq interface to assemble short and long transcriptome reads into transcripts using Oases.
- Transcriptomes: SOAPdenovo-Trans
SOAPdenovo-Trans (Luo et al., 2012) is a de novo transcriptome assembler adapted from the SOAPdenovo framework to resolve transcripts (and alternative splicing and different expression levels) from short read sequencing reads in the absence of any genomic reference. For more details, see the SOAPdenovo-Trans manual at "https://github.com/aquaskyline/SOAPdenovo-Trans".To run SOAPdenovo-Trans go to [De novo assembly → Assembly → Transcriptomes → SOAPdenovo-Trans]and follow Fig. 17.
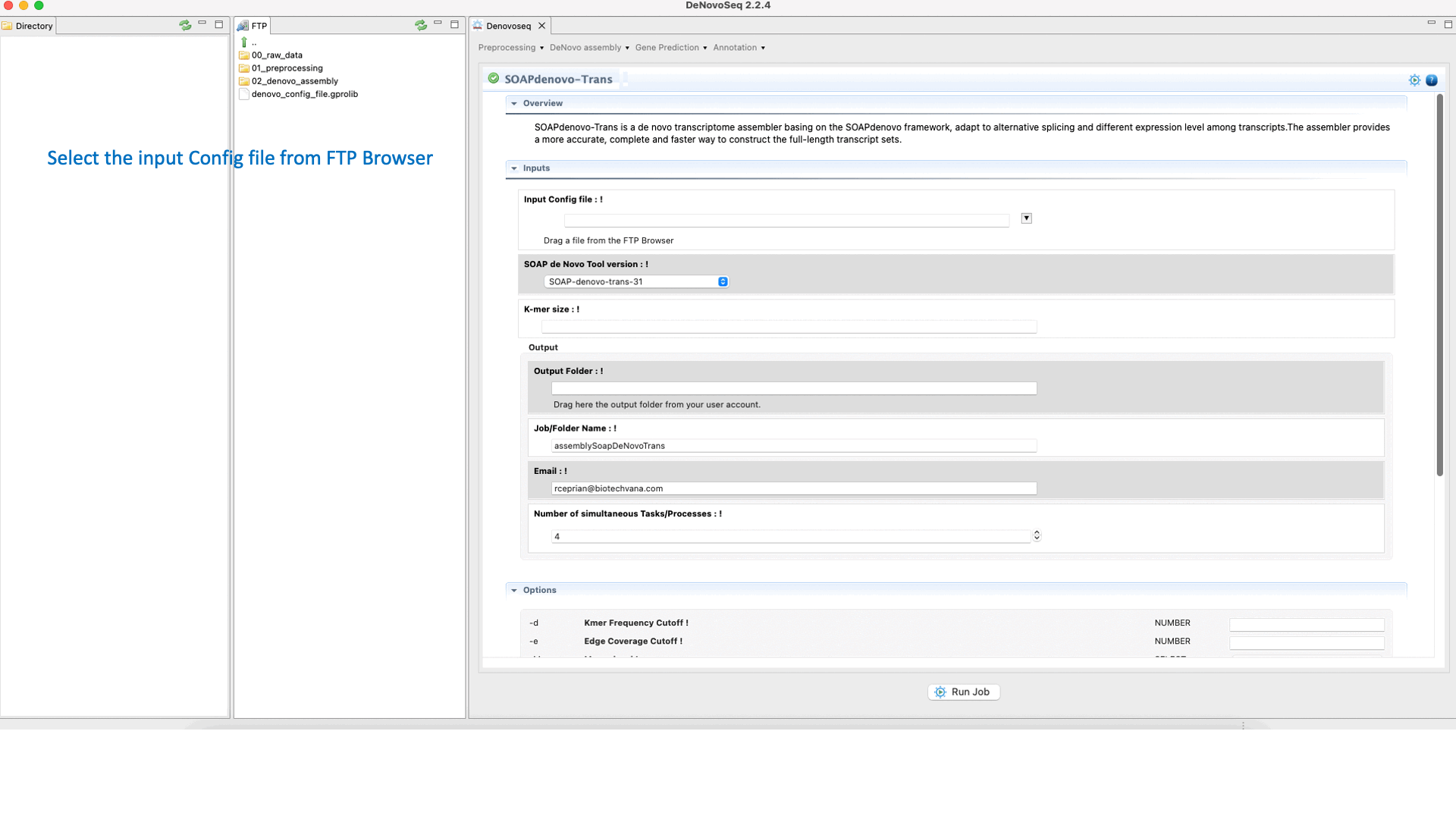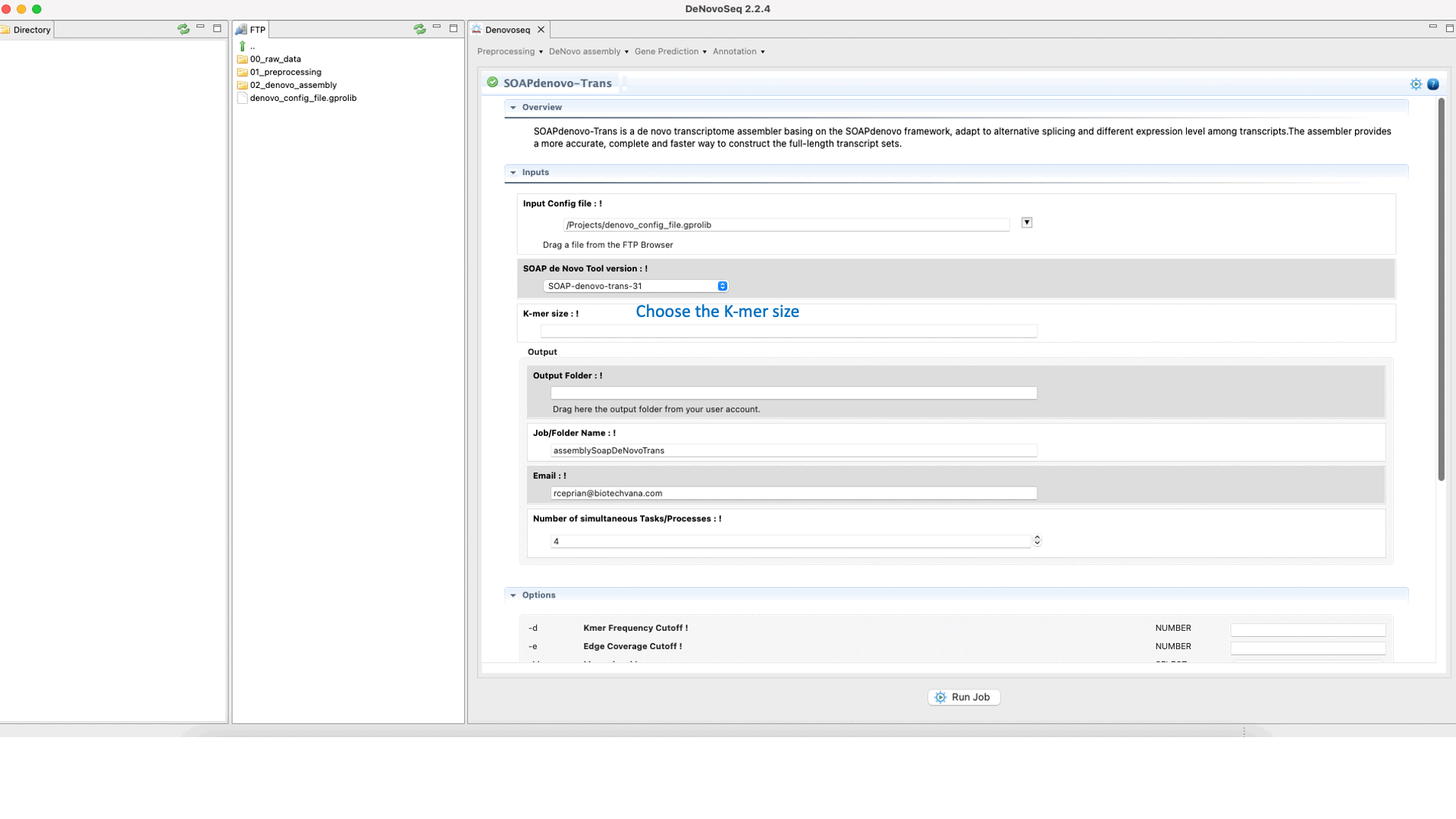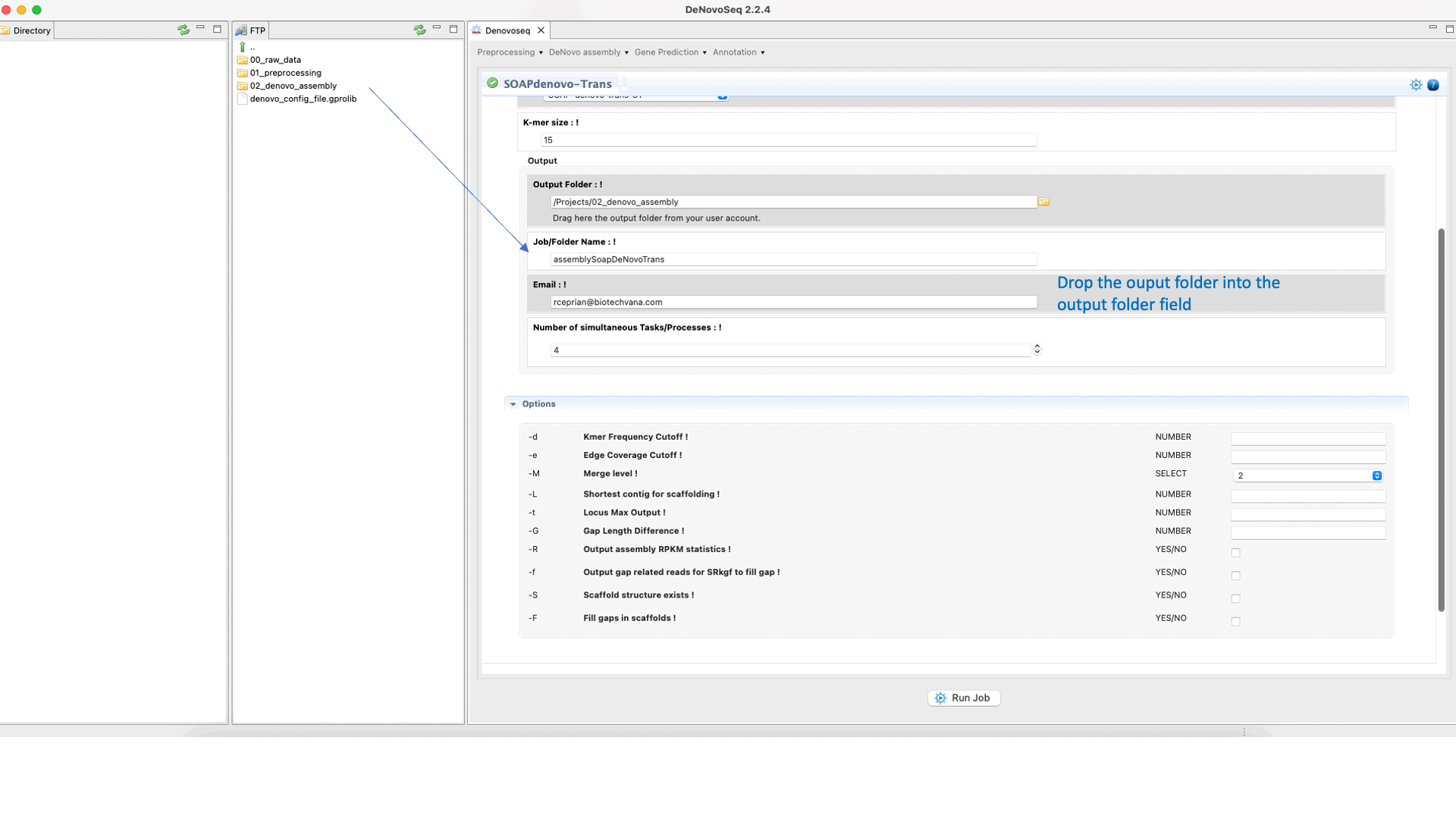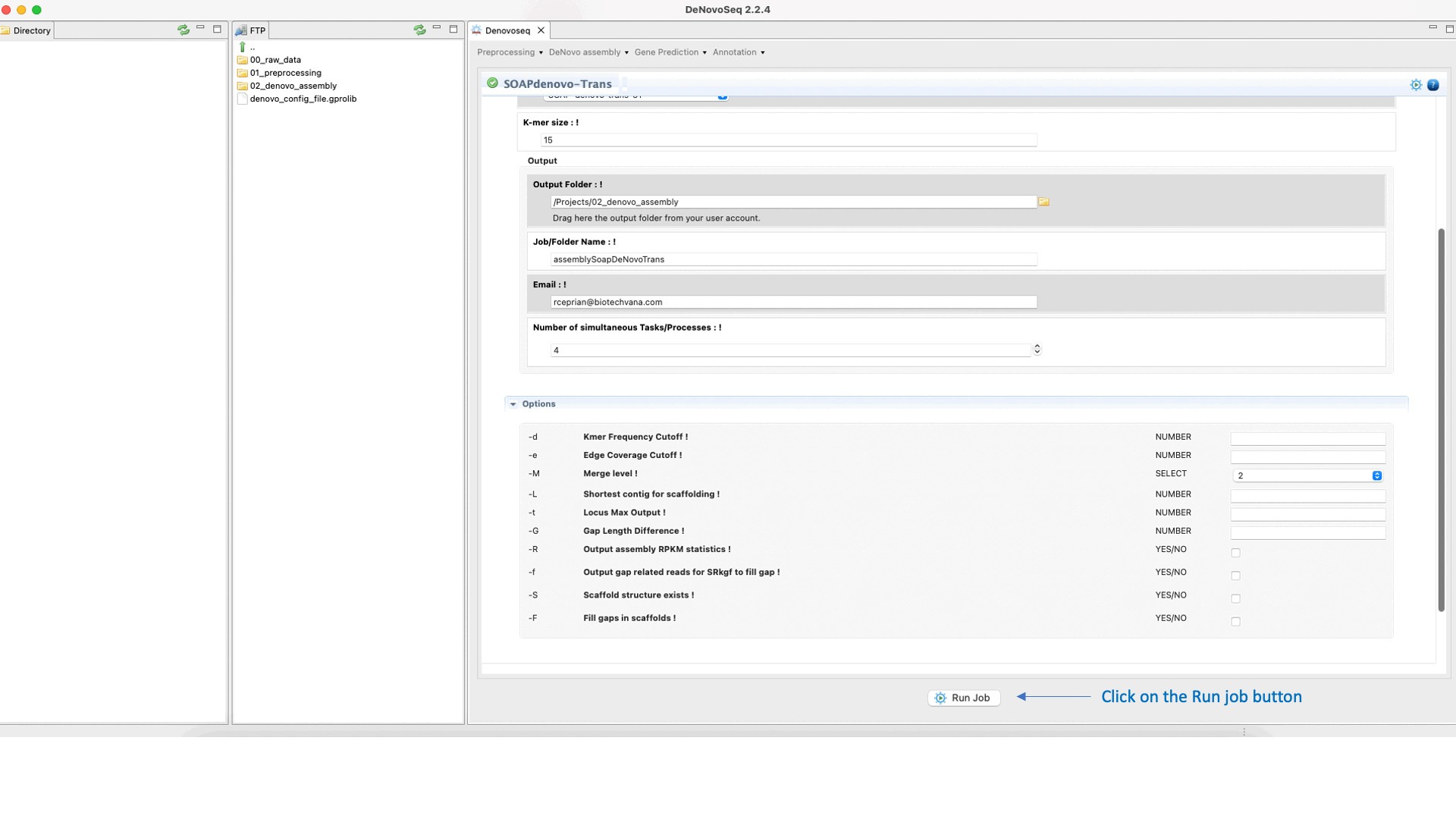
Figure 17: DeNovoSeq interface to assemble short transcriptome reads into transcripts using with SOAPdenovo-Trans.
- Genomes: Velvet
Velvet (Zerbino and Birney, 2008) ) is a de novo genome assembler that takes short read sequences and resolves high quality contigs. For more information, see the Velvet manual at "https://www.ebi.ac.uk/~zerbino/velvet/Manual.pdf".To run Velvet go to [De novo assembly → Assembly → Genomes → Velvet]and follow Fig. 18.
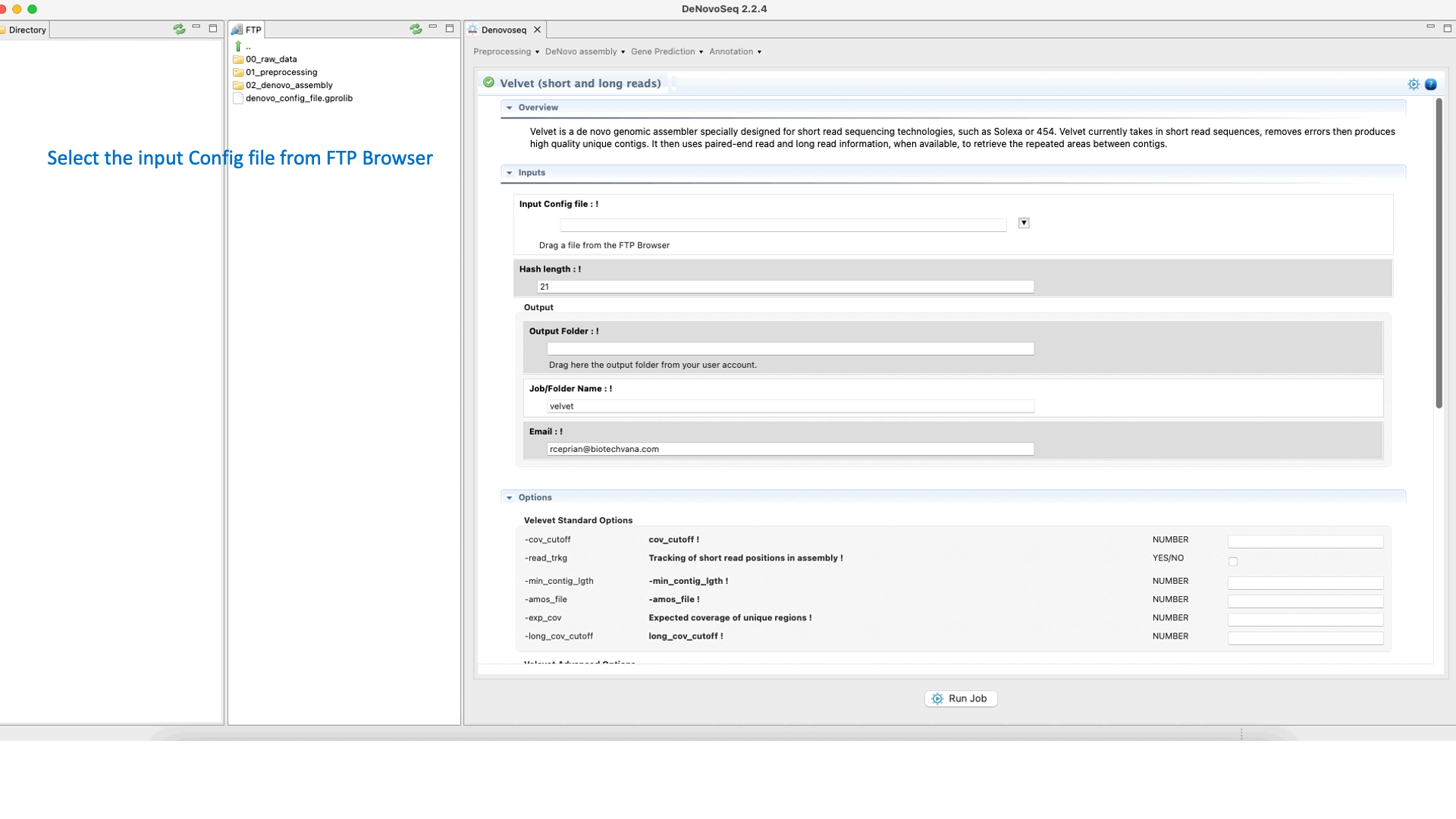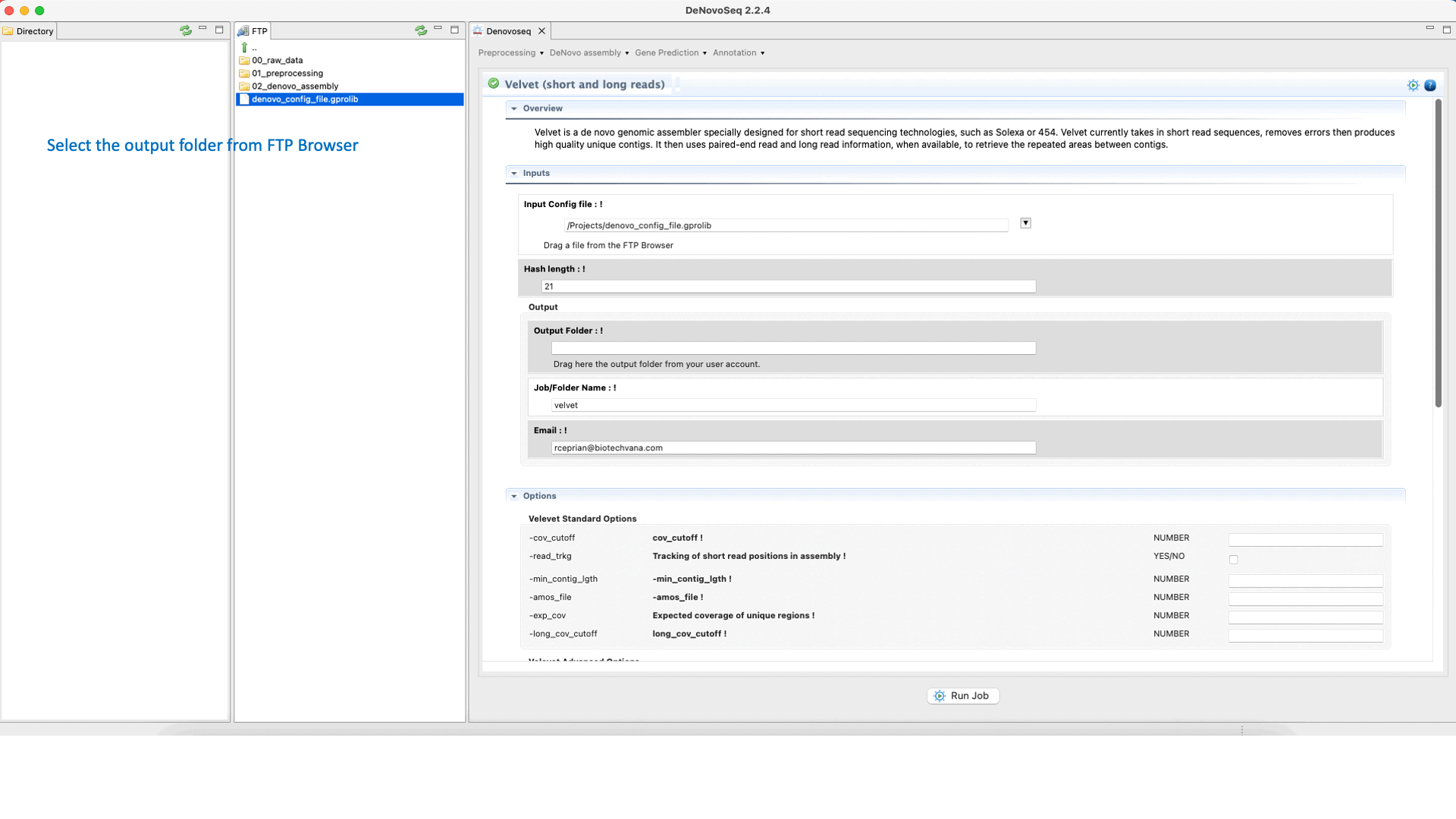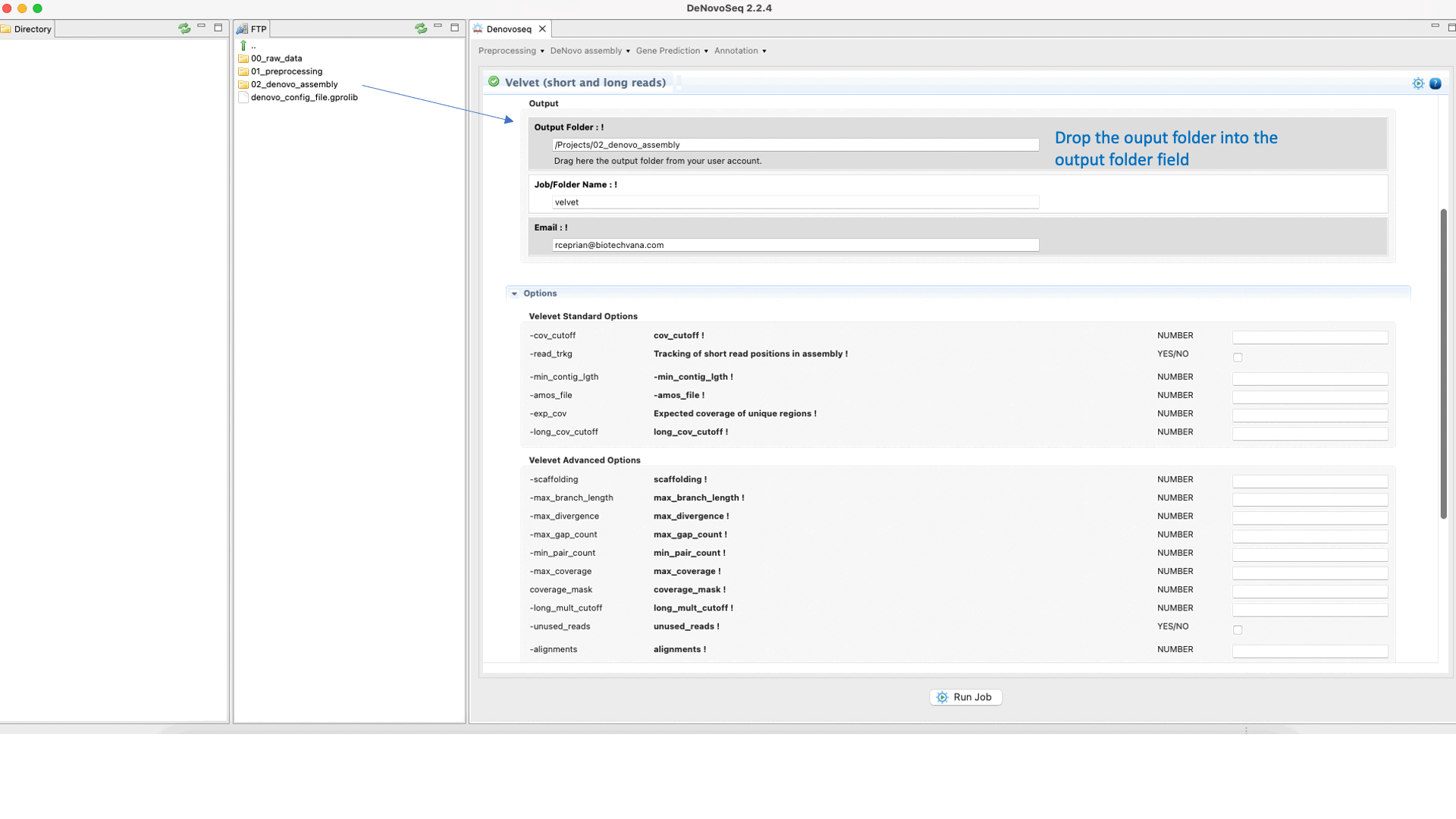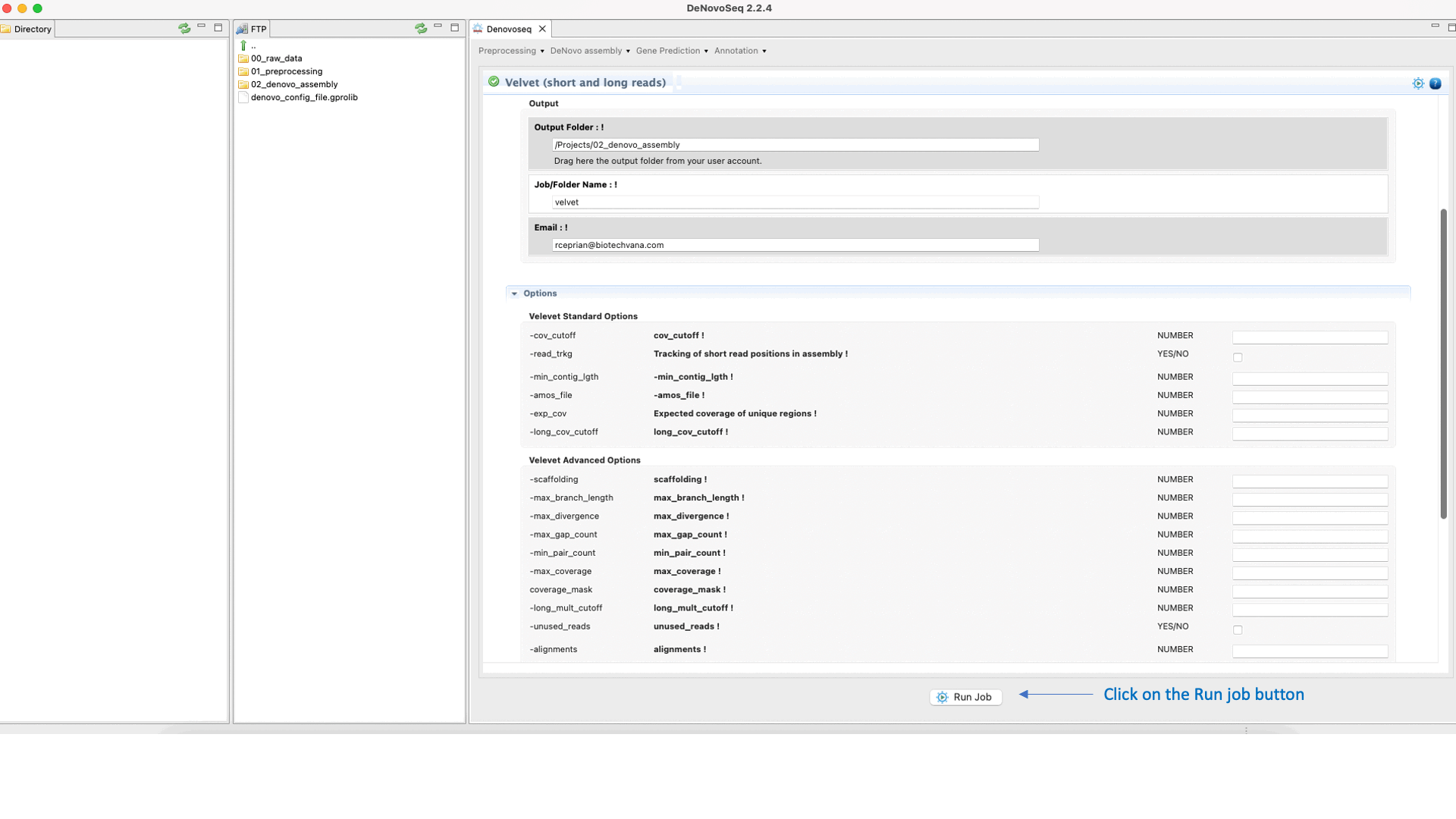
Figure 18: DeNovoSeq interface to assemble short and long genome reads into new contigs using Velvet.
- Genomes: SOAPdenovo2
SOAPdenovo2 (Luo et al., 2012) ) s an assembler designed to assemble Illumina GA short reads. SOAPdenovo reduces memory consumption in graph construction resolving repeat regions in contig assembly, increasing coverage and length in scaffold construction and improving gap closing. See the SOAPdenovo manual at "https://github.com/aquaskyline/SOAPdenovo2" for more information.To run SOAPdenovo2 go to [De novo assembly → Assembly → Genomes → SOAPdenovo2]and follow Fig. 19.
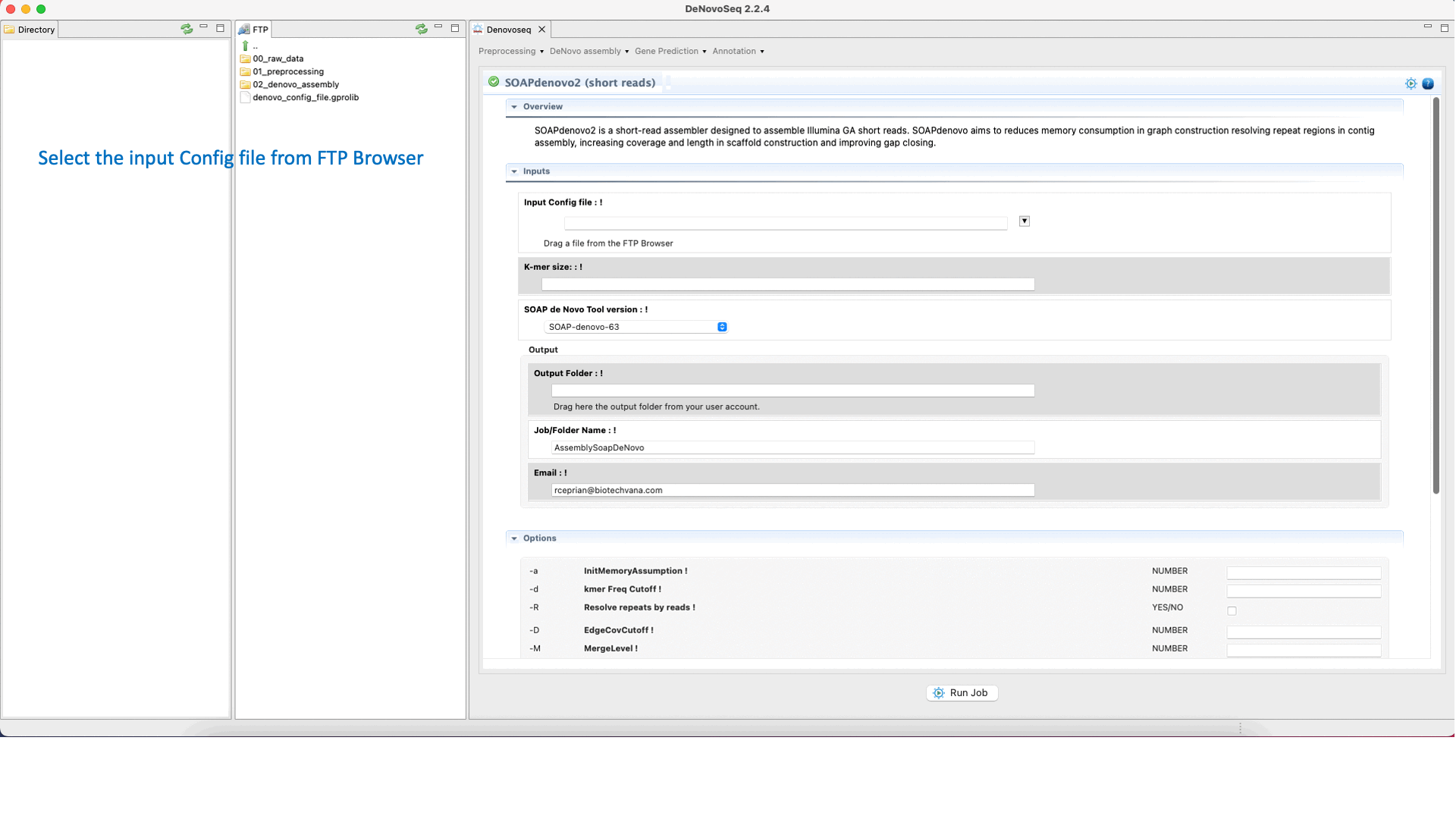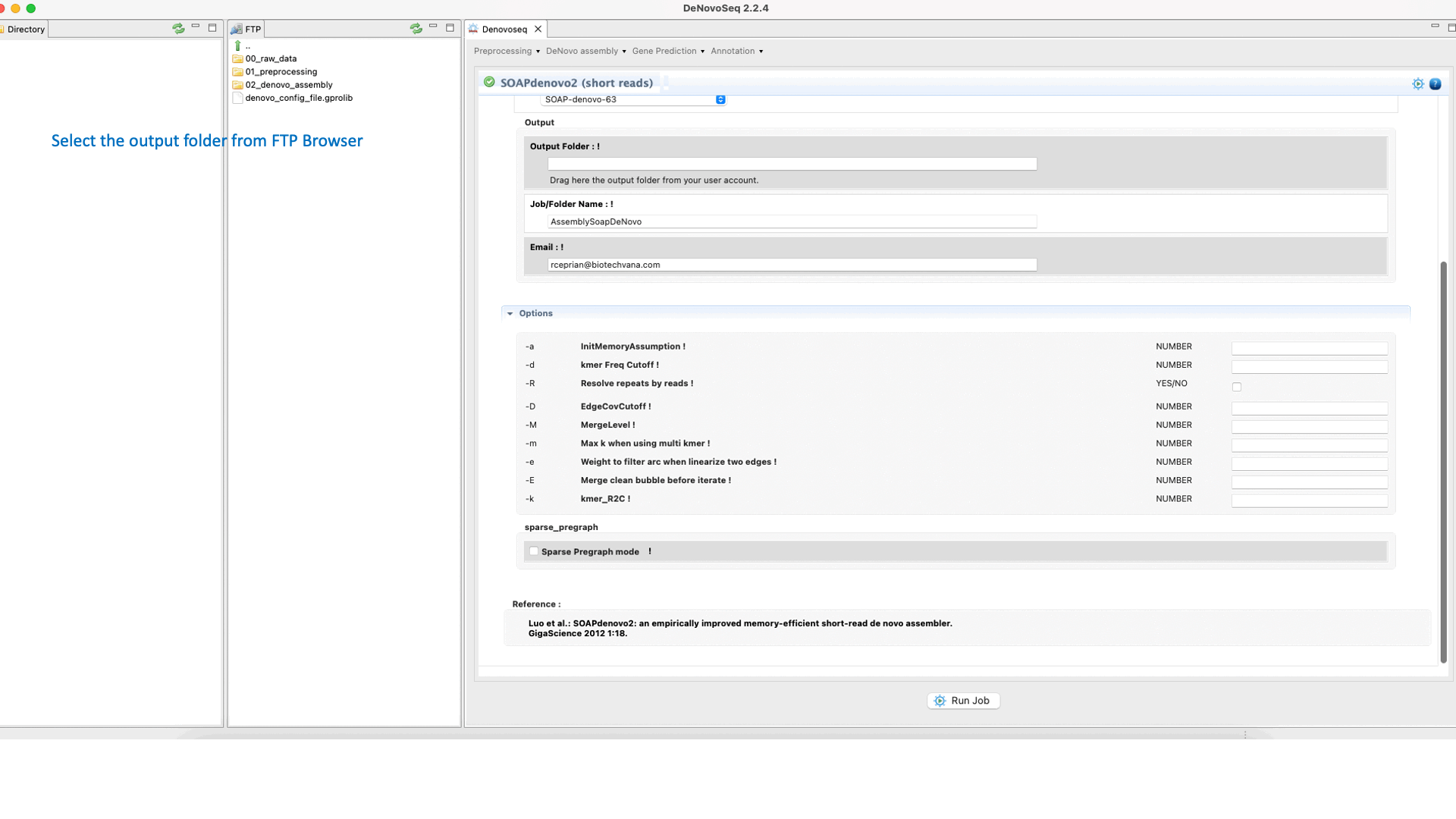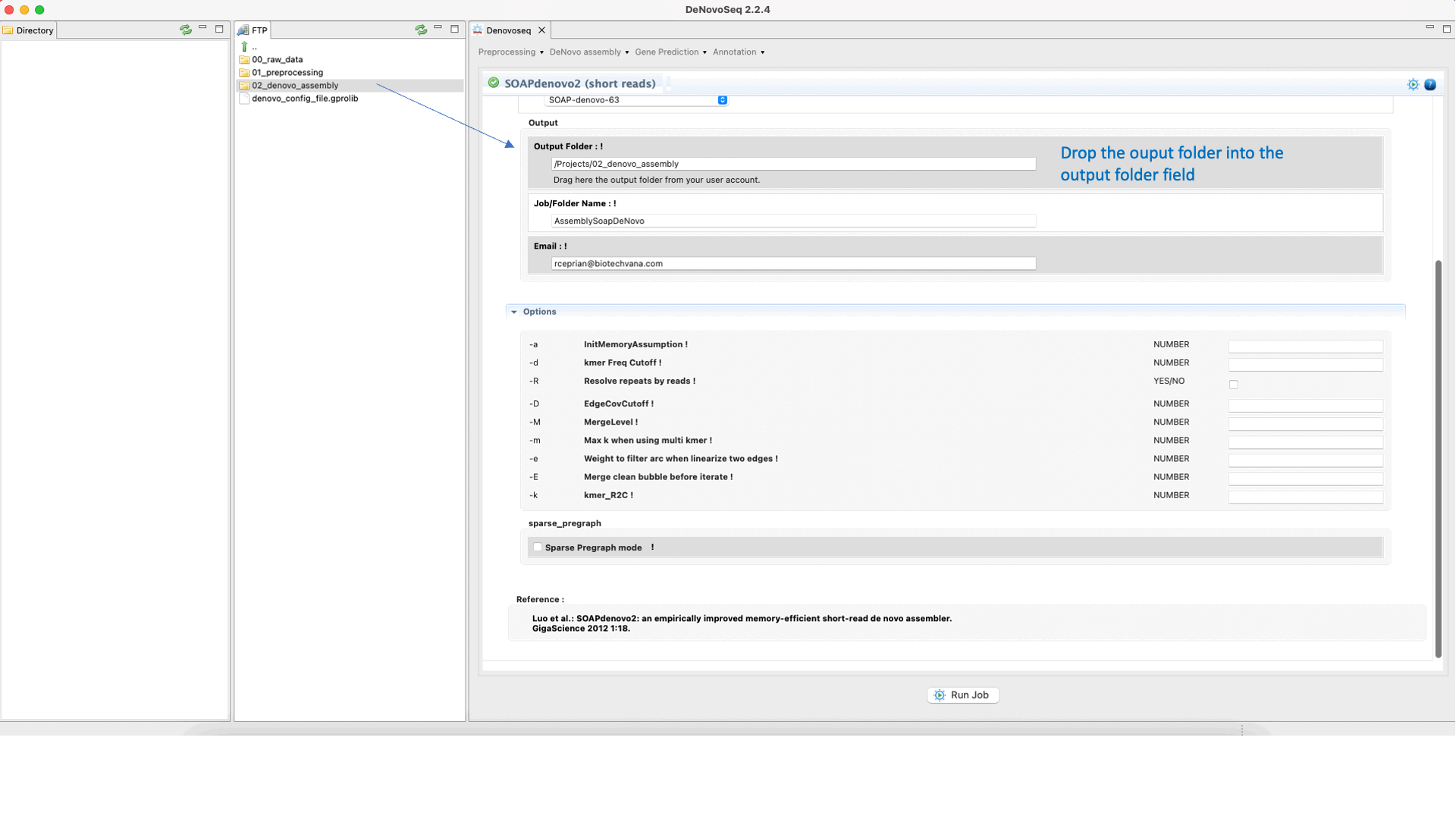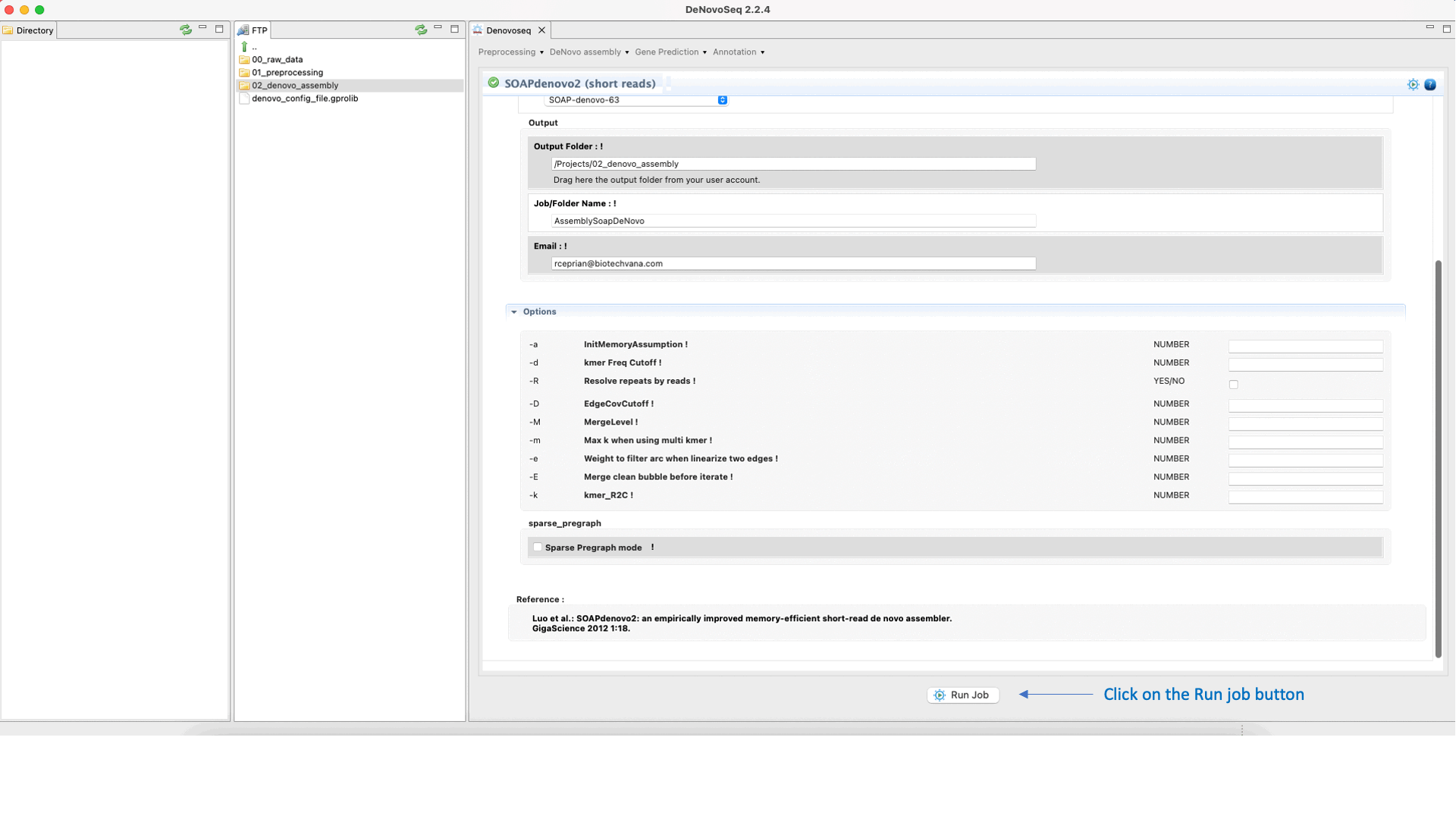
Figure 19: DeNovoSeq interface to assemble short genome reads into contigs using SOAPdenovo2.
- Genomes: CANU
CANU (Koren et al., 2017) is an assembler of the Celera Assembler designed for high-noise single-molecule sequencing such as the PacBio RSII or Oxford Nanopore MinION. CANU is a hierarchical assembly pipeline, which runs in four steps:To run CANU go to [De novo assembly → Assembly → Genomes → CANU]and follow Fig. 20.
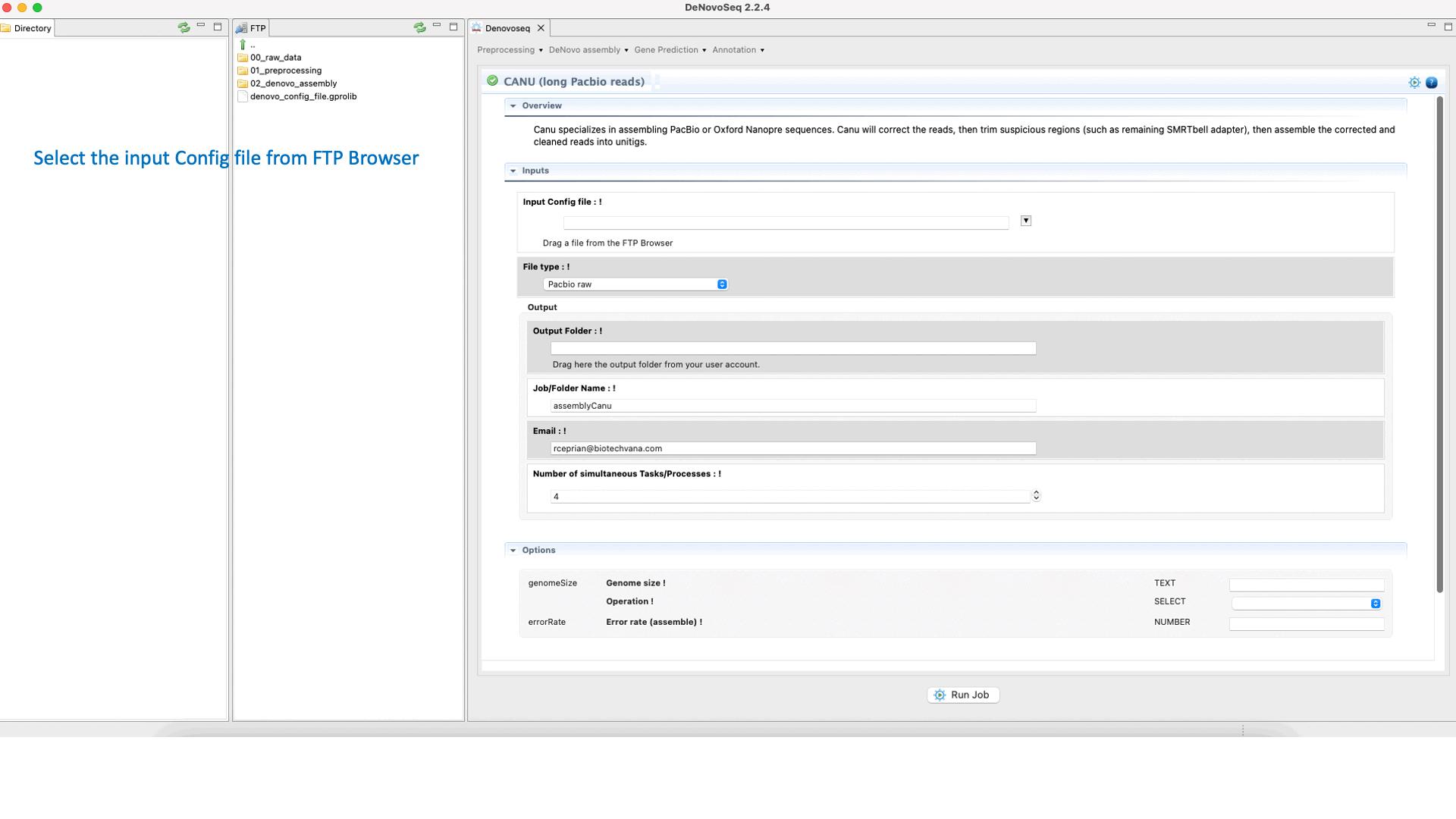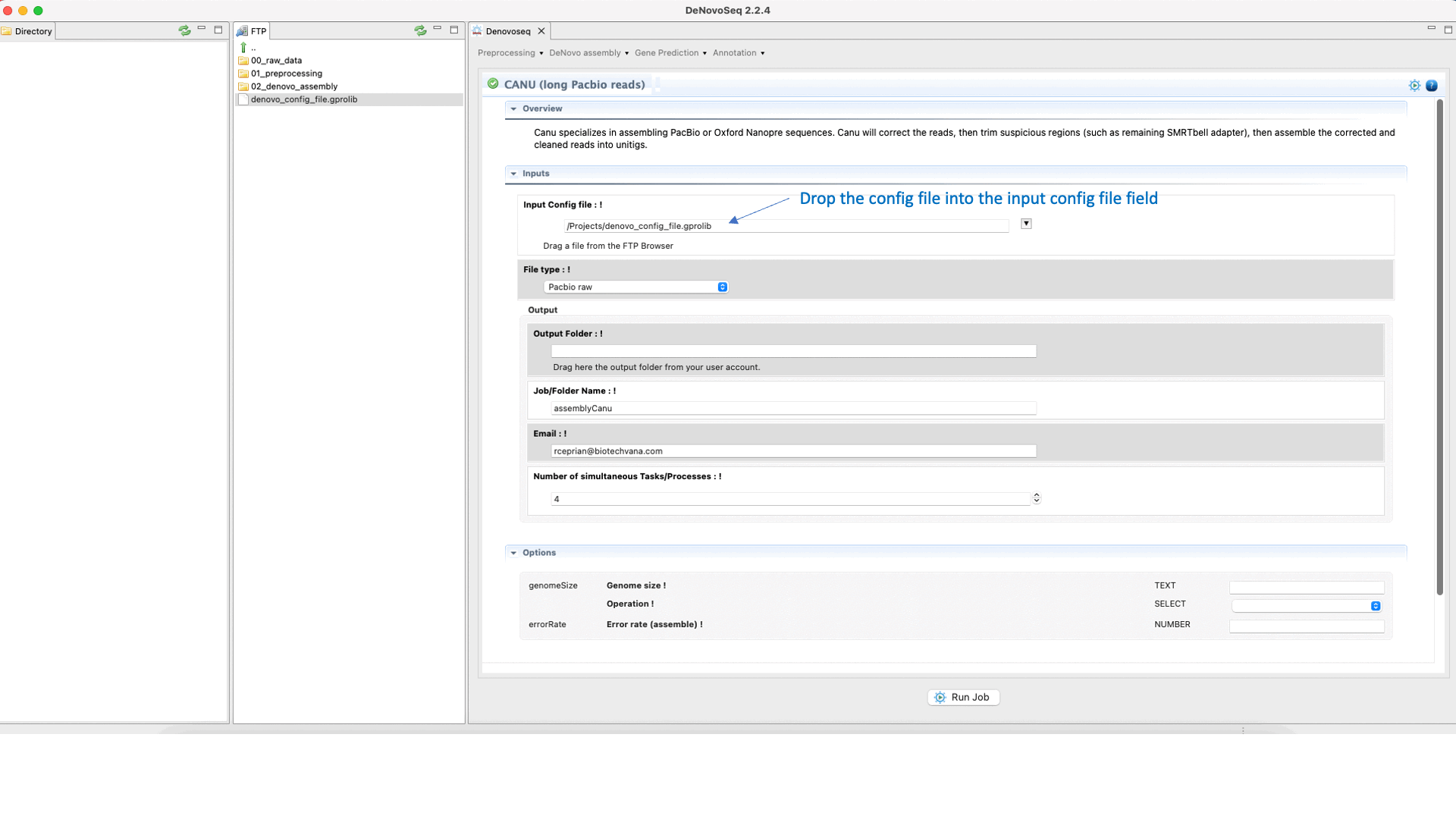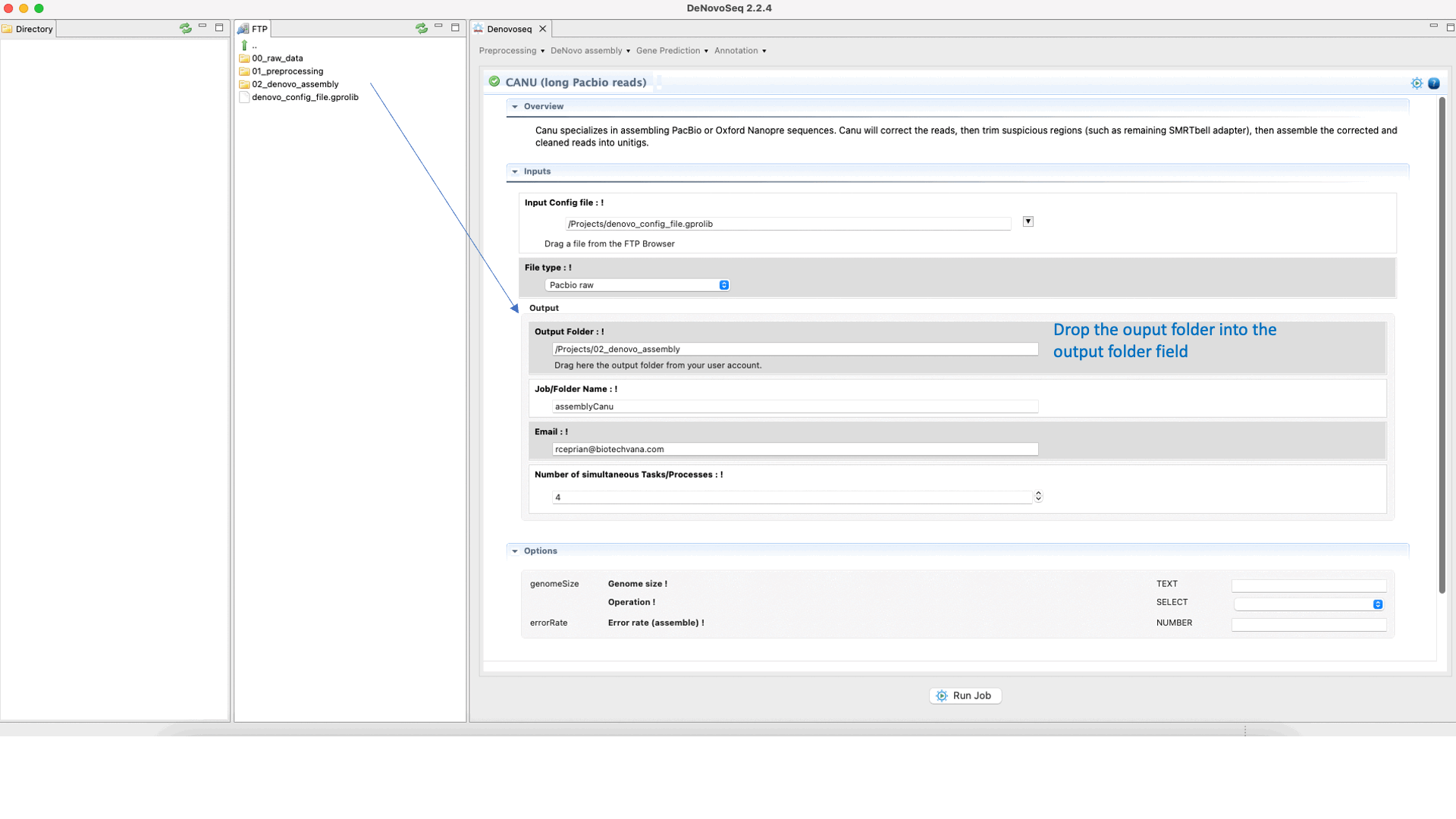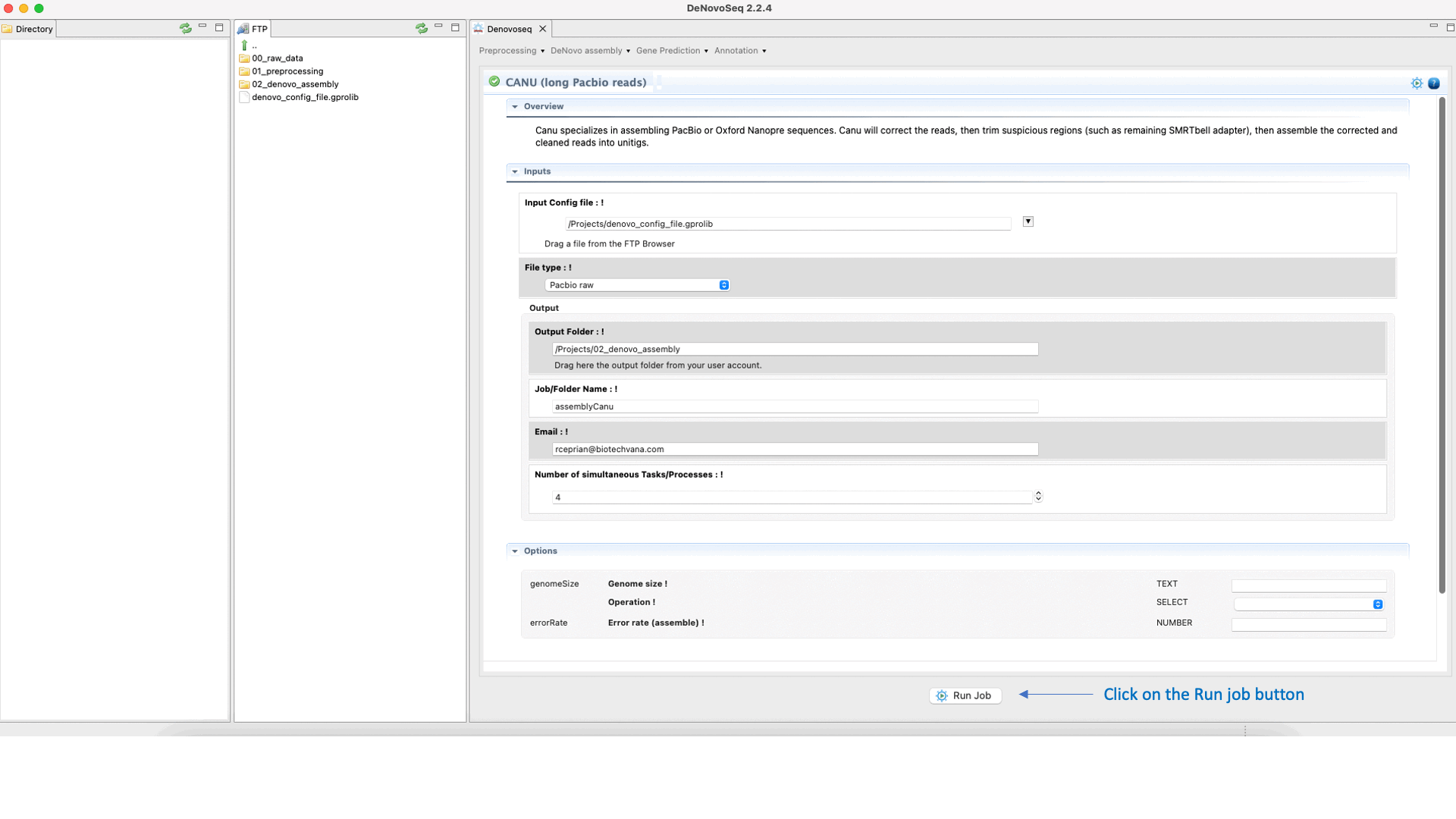
Figure 20: DeNovoSeq interface to assemble long genome reads into contigs using CANU.
- Genomes: SPAdes
SPAdes (Bankevich et al., 2012) is an assembler recommended to reconstruct small genomes (bacterial fungal and others). SPAdes supports paired-end reads, mate-pairs and unpaired reads. See the SPAdes manual at "http://cab.spbu.ru/software/spades/ " for more information.To run SPAdes go to [De novo assembly → Assembly → Genomes → SPAdes]and follow Fig. 21.
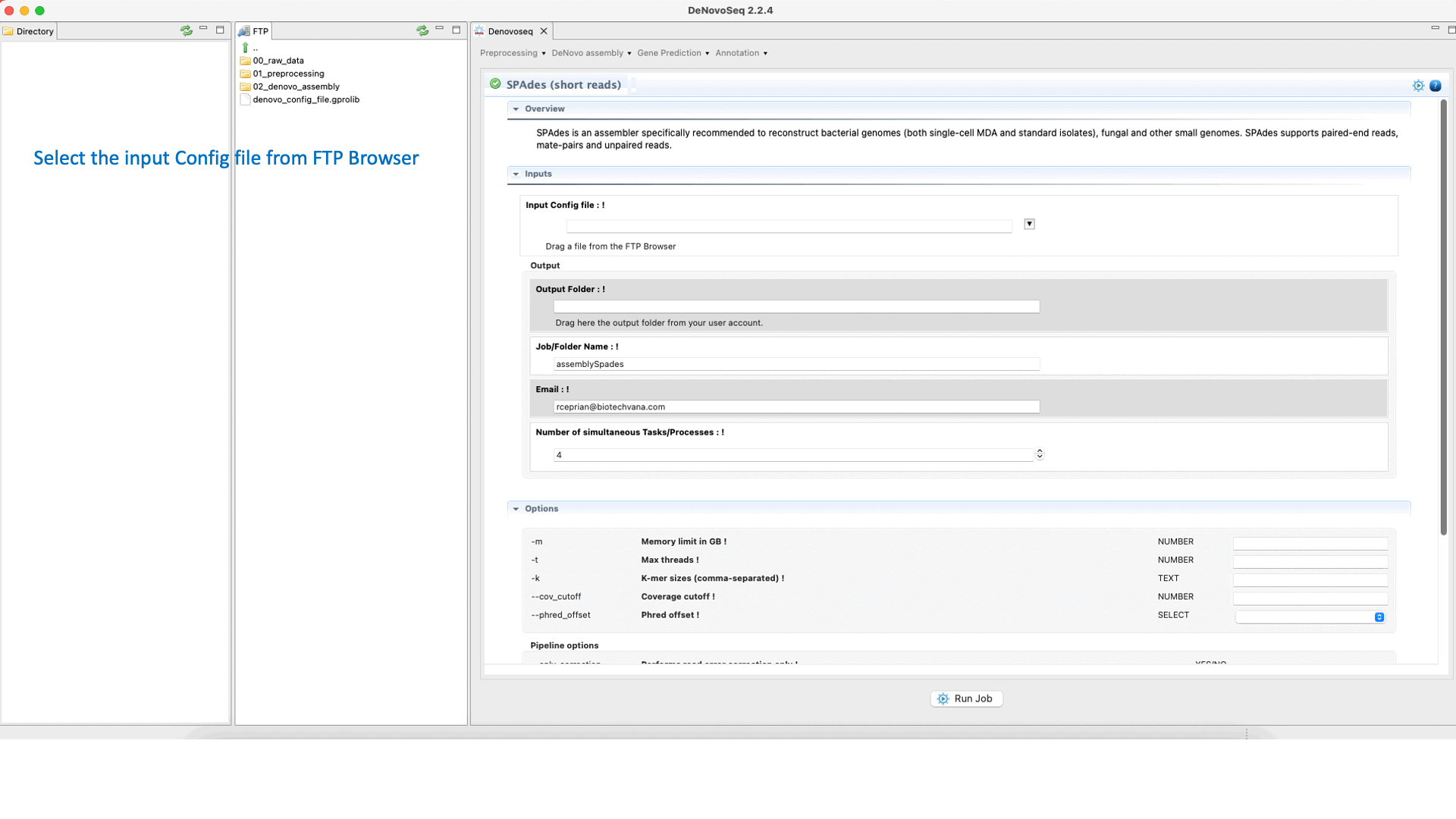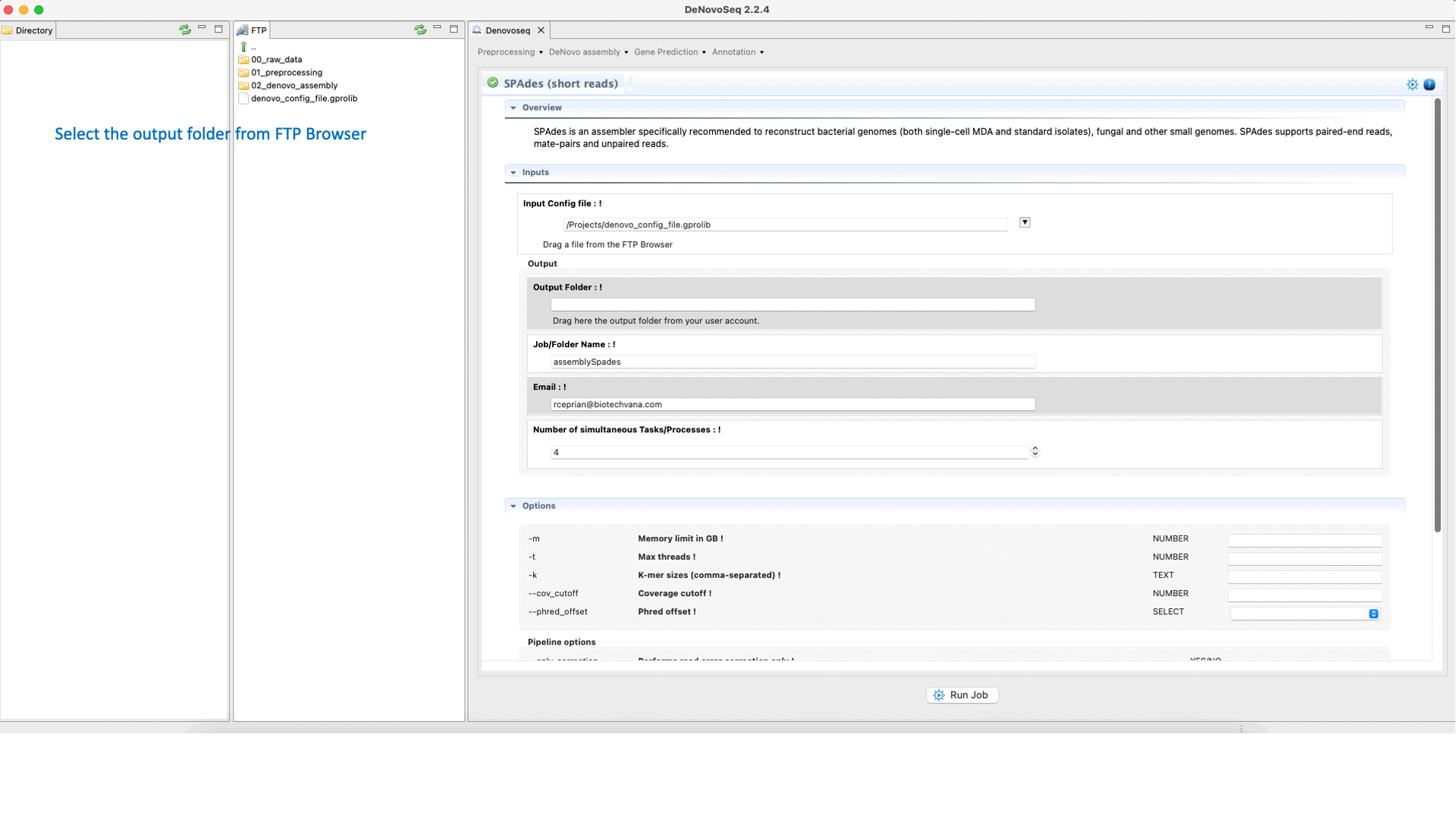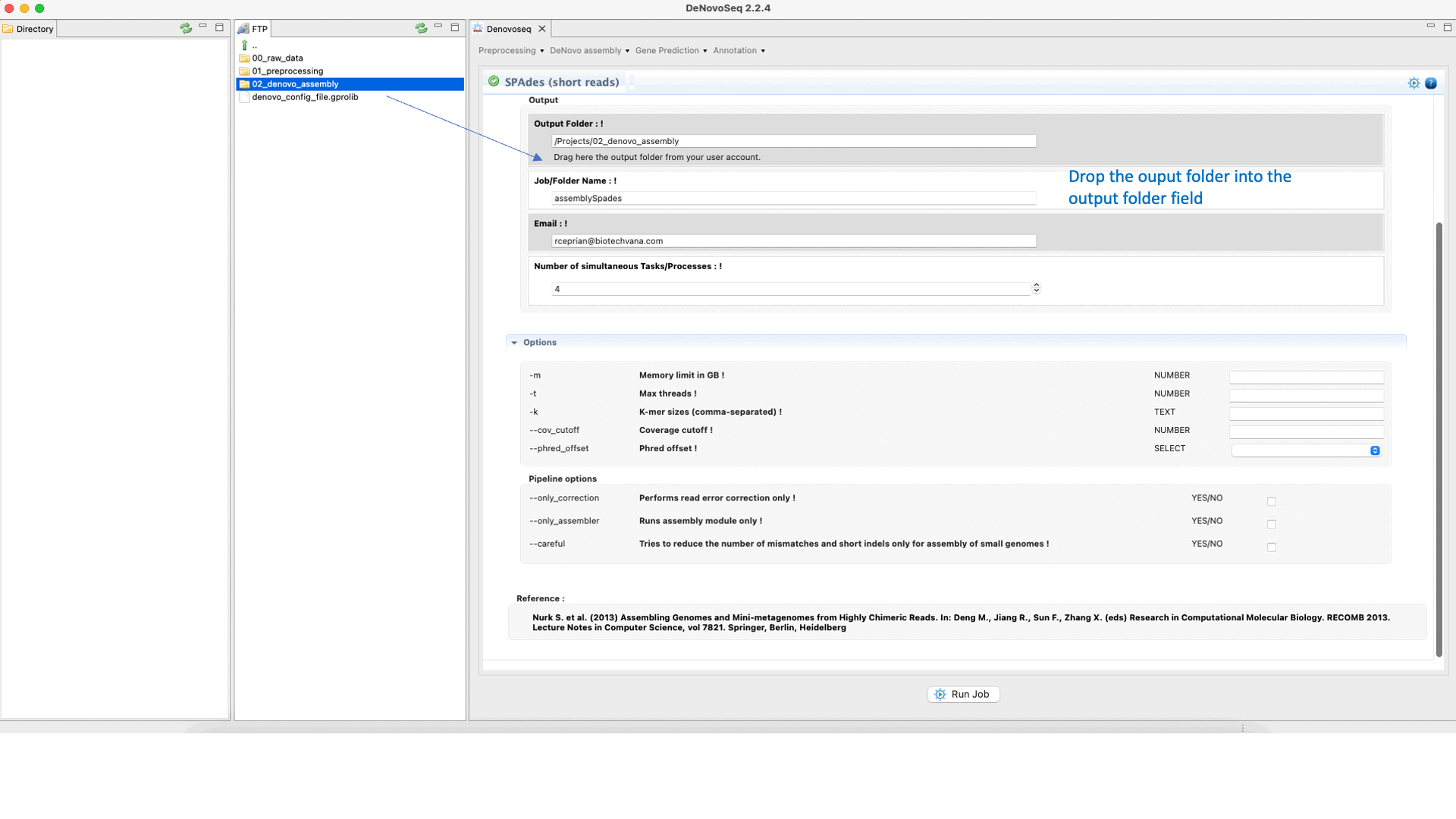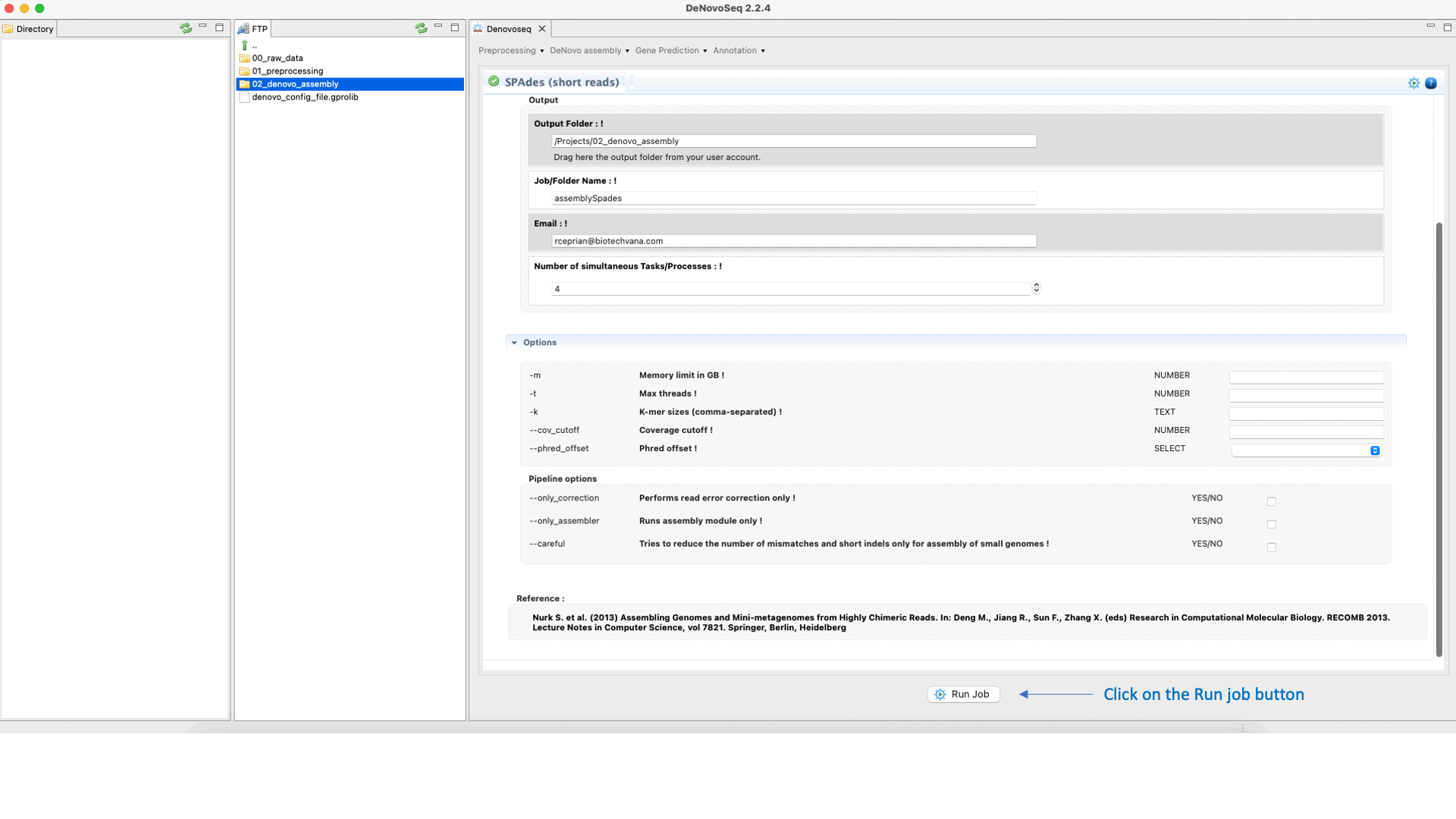
Figure 21: DeNovoSeq interface to assemble bacterial genome reads into new contigs using SPAdes.
- Gap filling: GapCloser
GapCloser (Luo et al., 2012) is a tool that closes gaps using the abundant pair-to-pair relationship of short reads. See the GapCloser manual at "https://vcru.wisc.edu/simonlab/bioinformatics/programs/soap/GapCloser_Manual.pdf " for more information.To run GapCloser go to [De novo assembly → Gap filling → GapCloser]and follow Fig. 22.
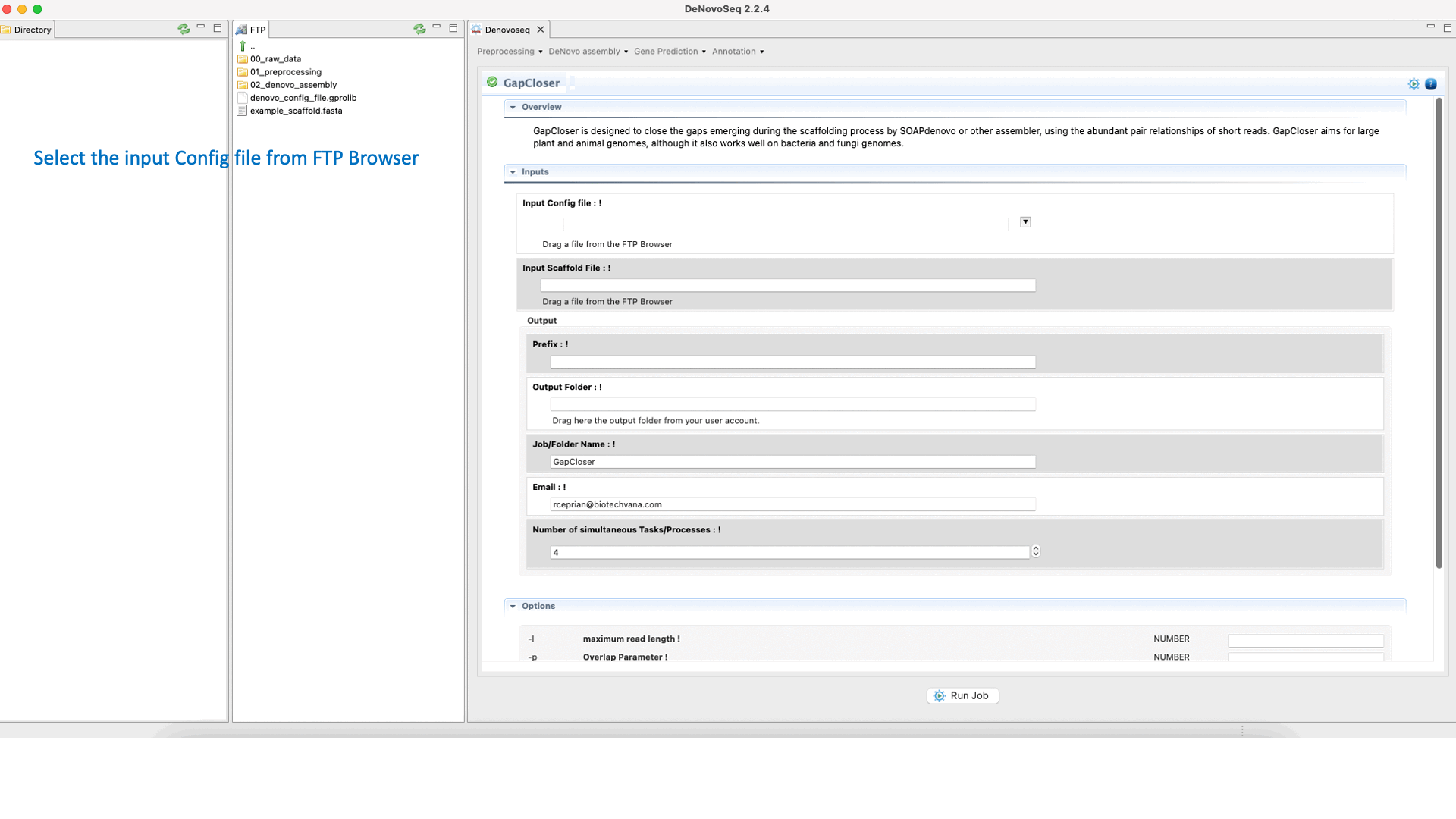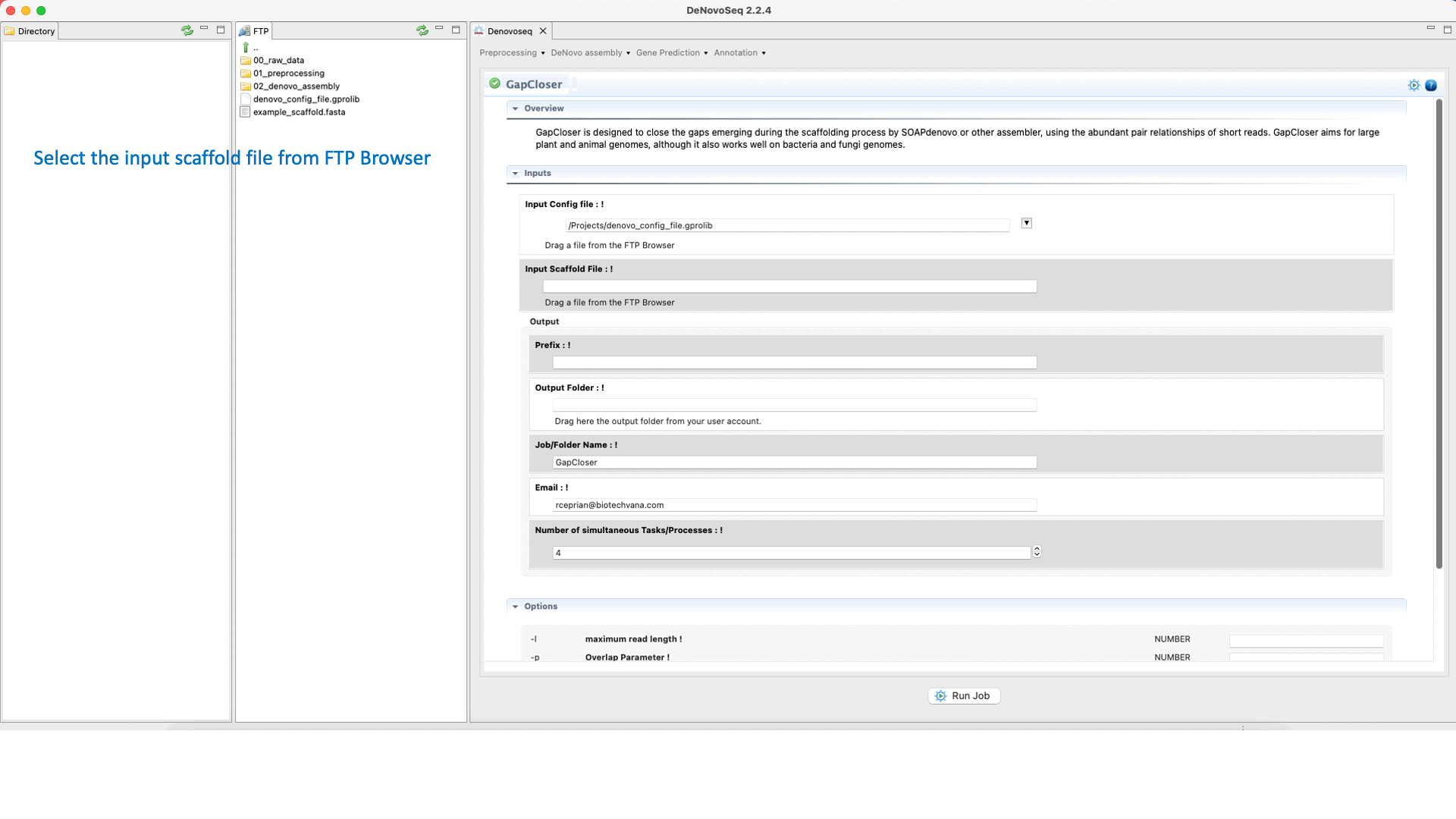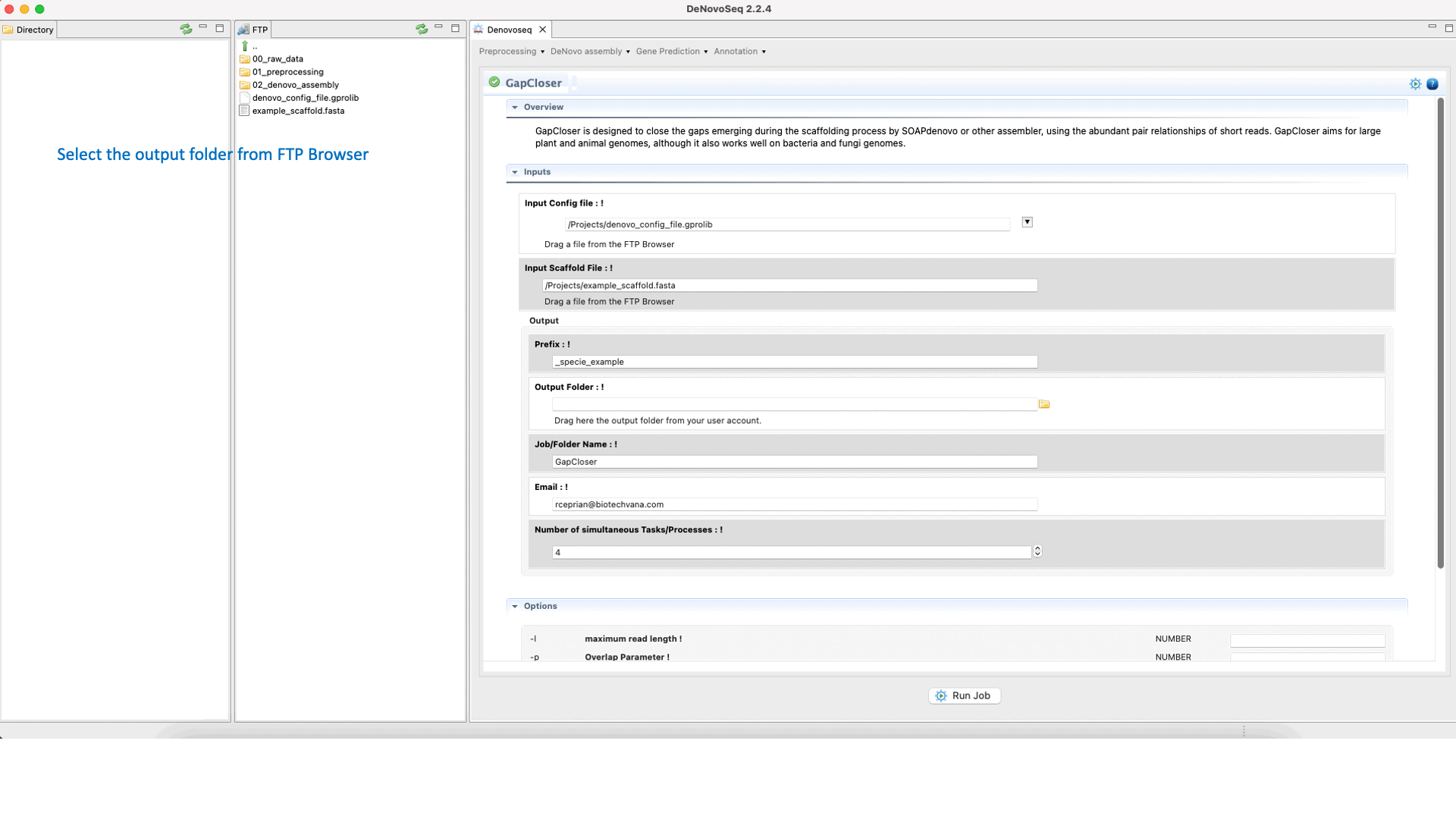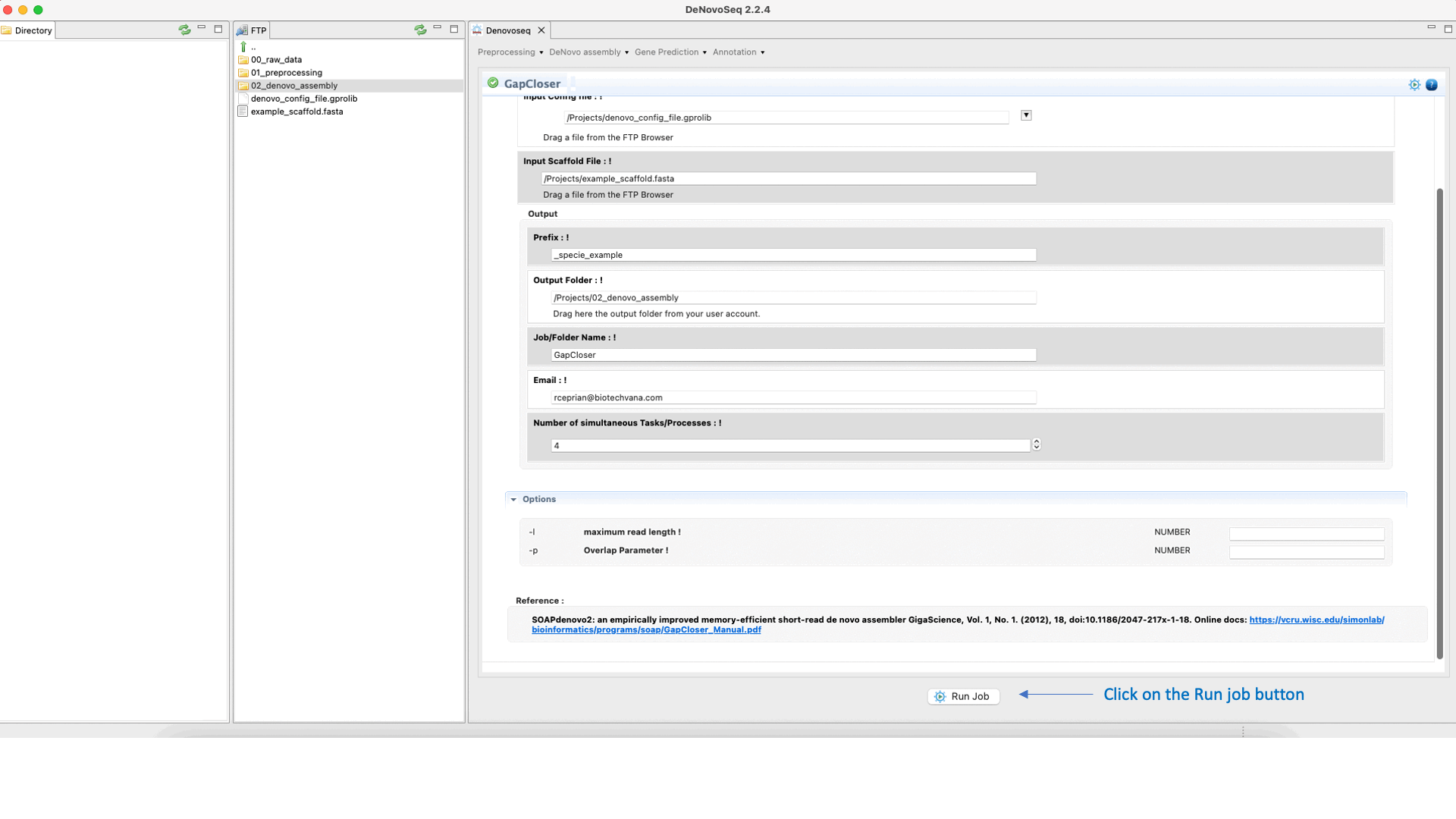
Figure 22: DeNovoSeq interface for GapCloser.
Results delivered by de novo assemblies are usually fragmented sets of genomic sequences (contigs) that can be re-ordered, edited and joined using the paired-end information in larger sequences called scaffolds. DeNovoSeq implements interfaces for two alternative scaffolders; BESST (Sahlin et al 2014) and OPERA (Gao et al., 2011).
- Scaffolding: BESST
BESST (Sahlin et al 2014) is a scaffolder that includes several tools to build a “contig graph” from available assembly information, obtaining scaffolds from this graph and accurate gap size information. See the BESST manual at "https://github.com/ksahlin/BESST" for more information.To run BESST go to [De novo assembly → Scaffolding → BESST]and follow Fig. 23.
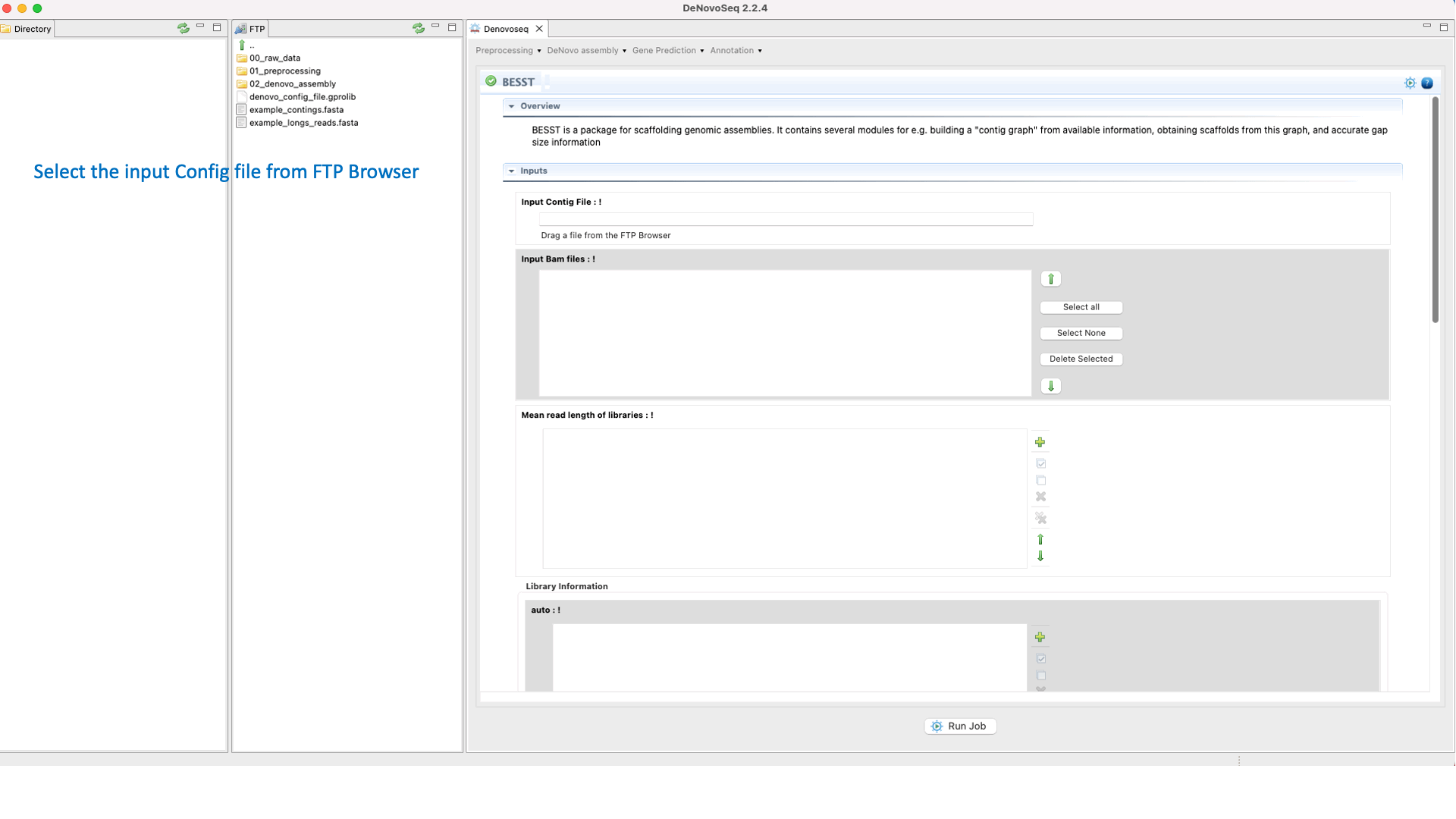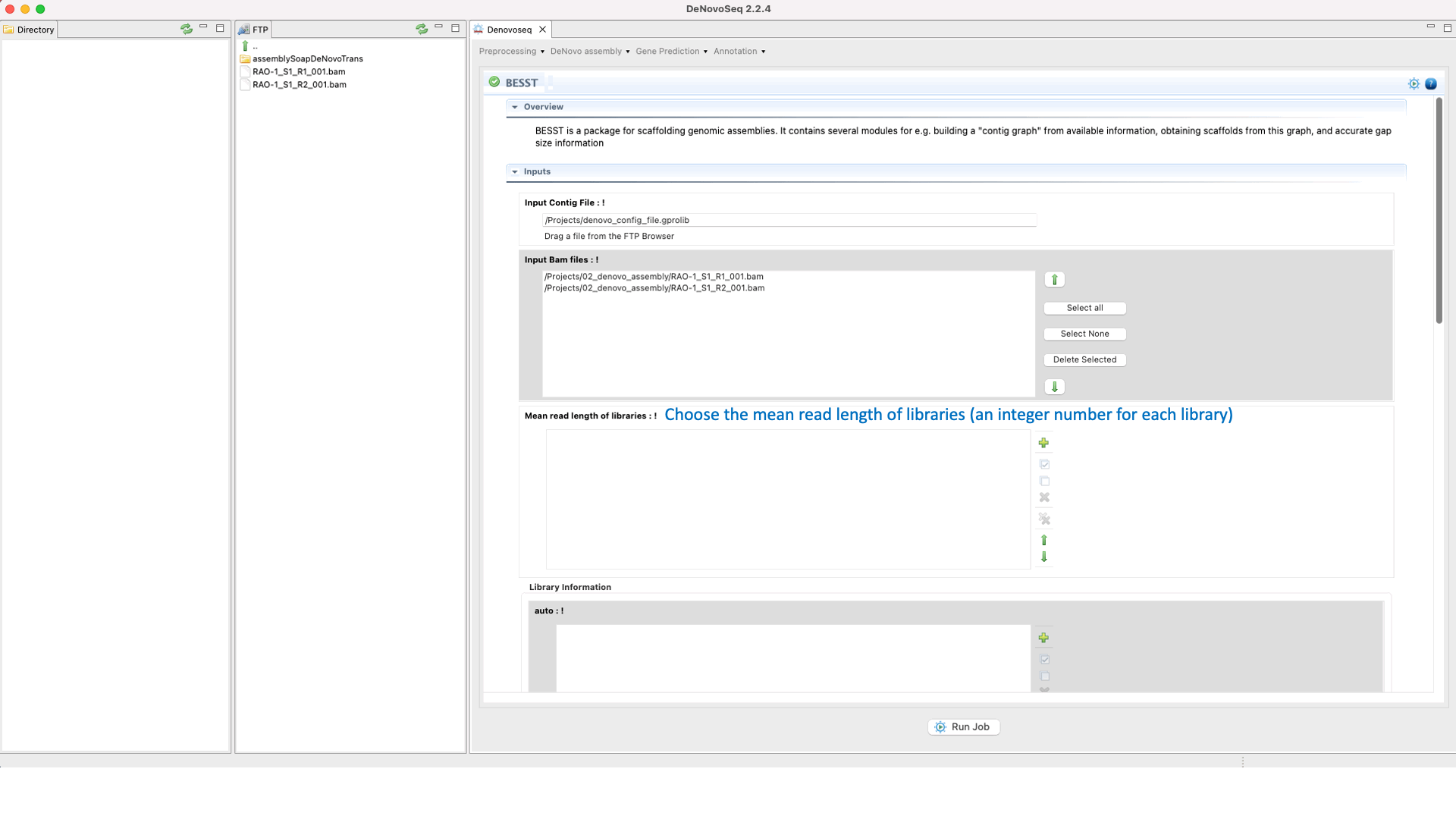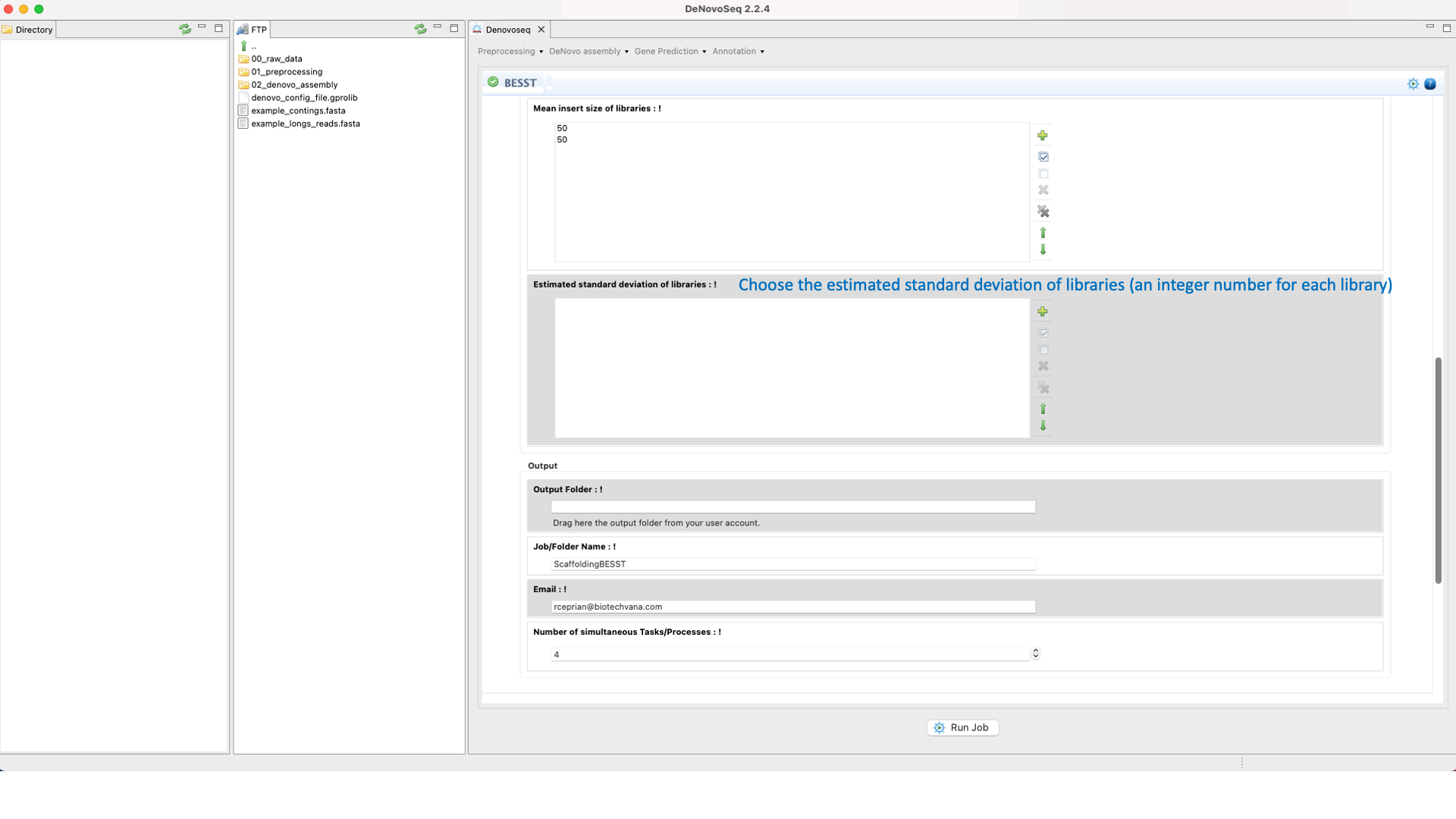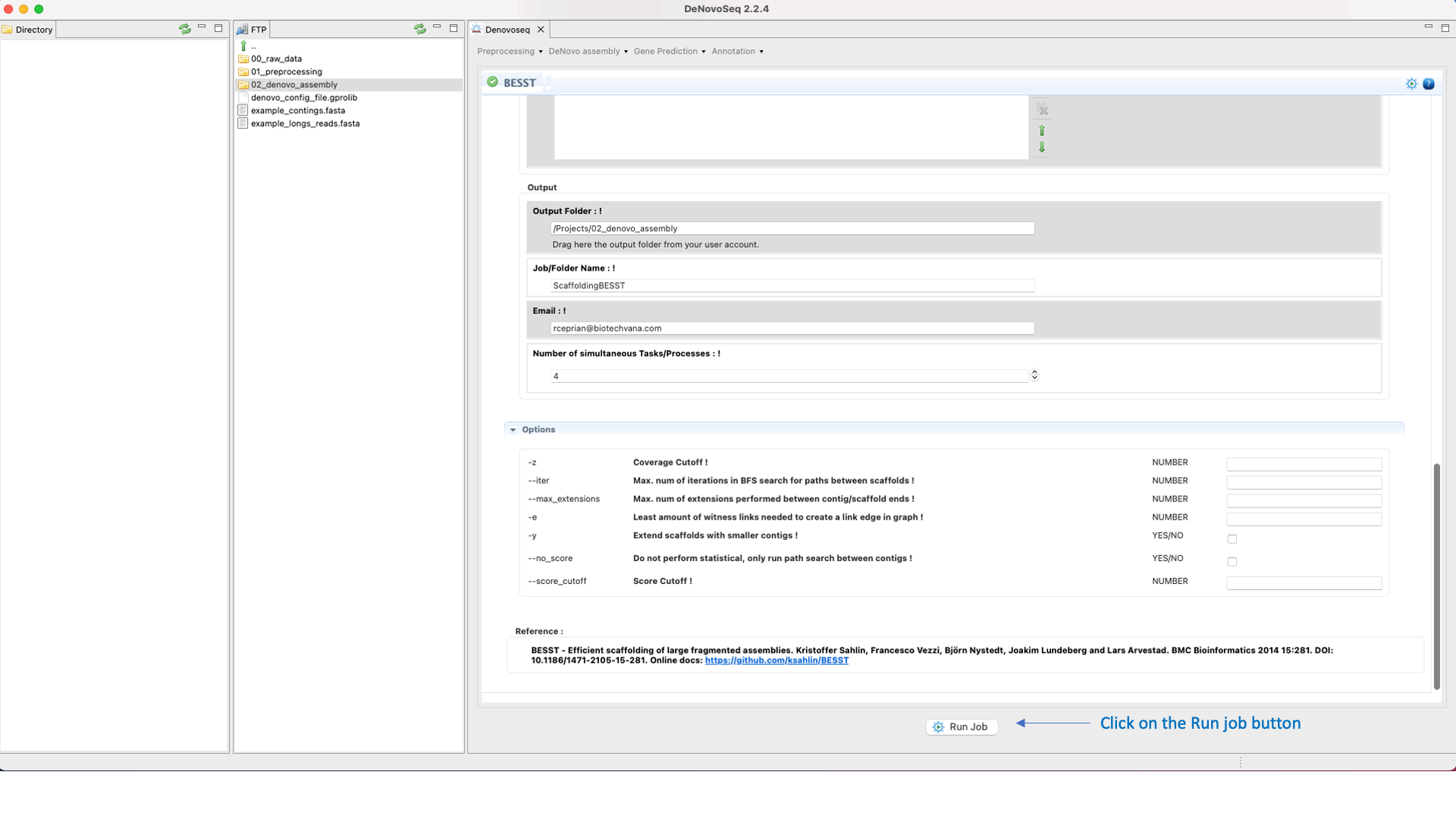
Figure 23: DeNovoseq interface to manage BESST scaffolder.
- Scaffolding: OPERA-LG long reads
OPERA (Gao et al., 2011) is a scaffolder that uses information from paired-end/mate-pair/long reads to order and orient the intermediate contigs/scaffolds assembled in a genome assembly project. See the OPERA manual at "https://sourceforge.net/projects/operasf/files/OPERA-LG%20version%202.0.6/" for more information.To run OPERA go to [De novo assembly → Scaffolding → OPERA-LG long reads]and follow Fig. 24.
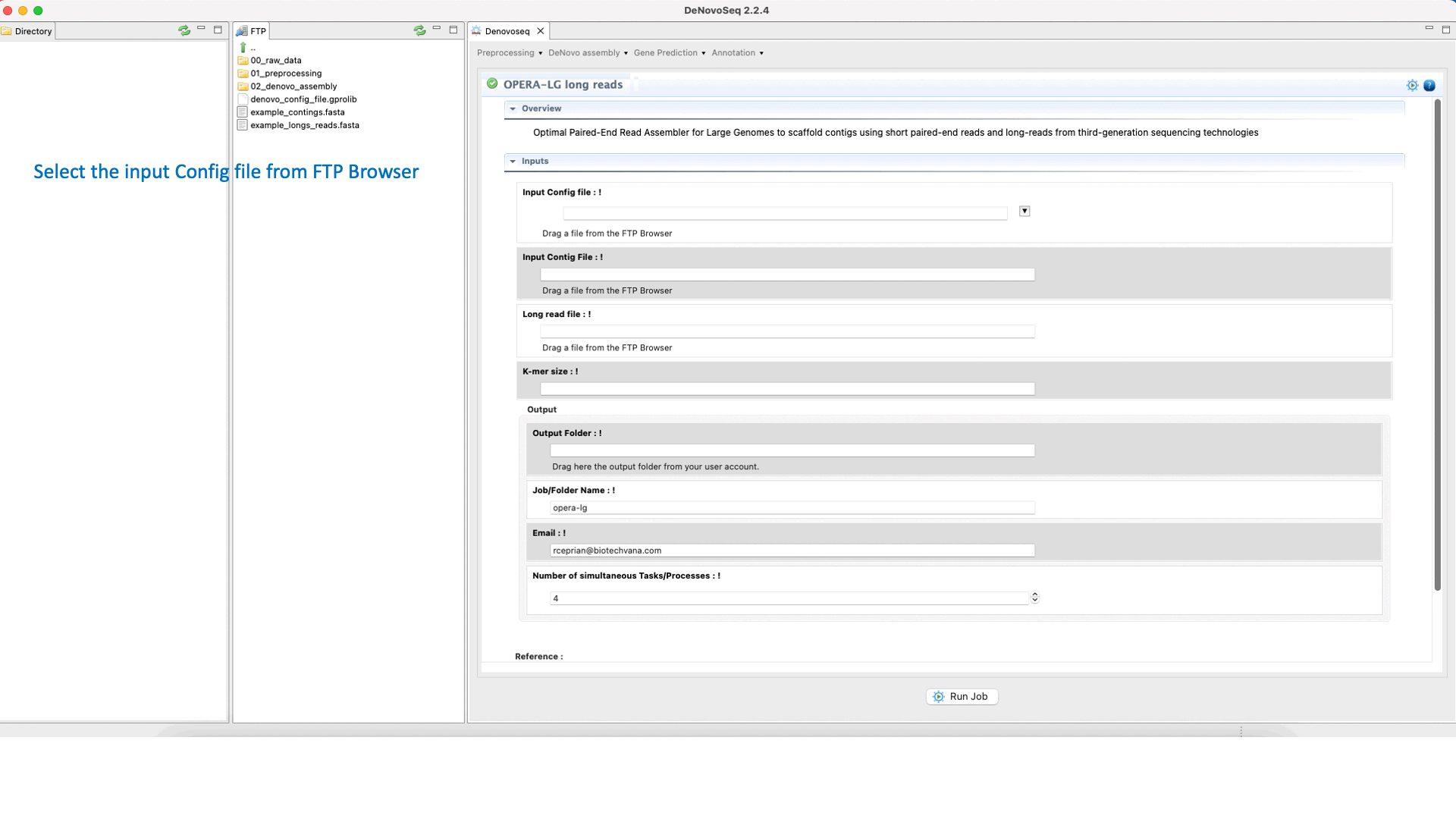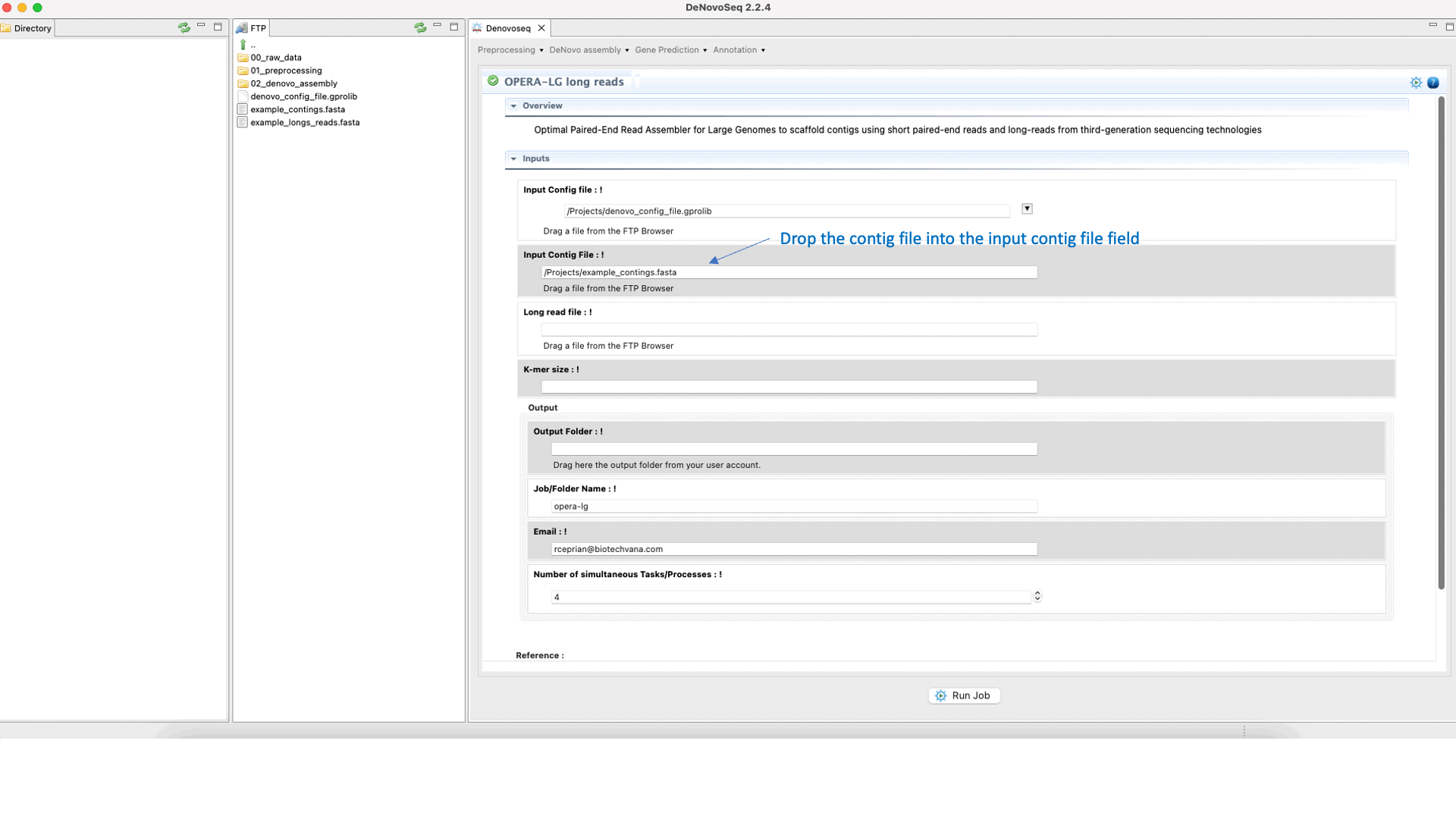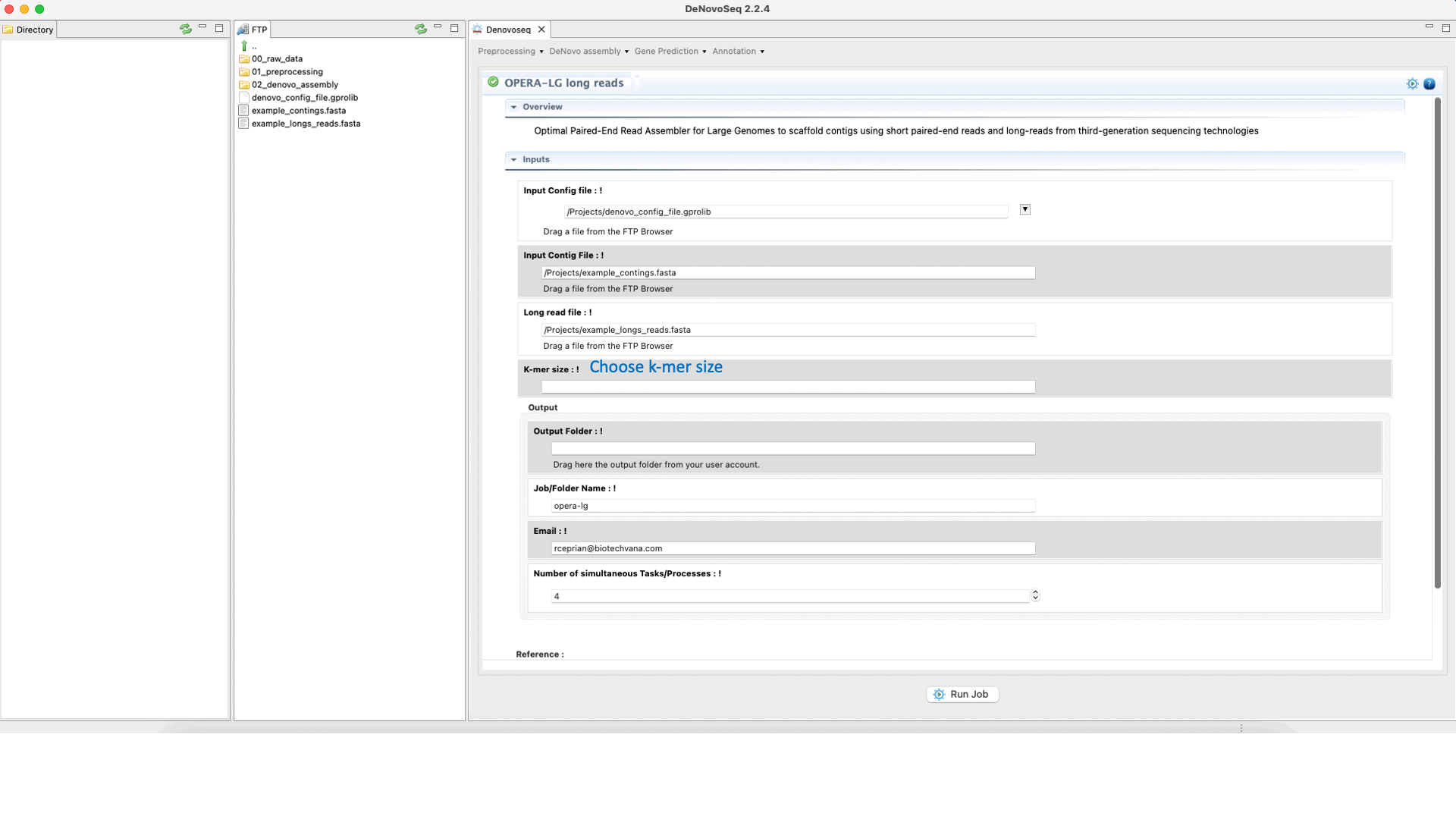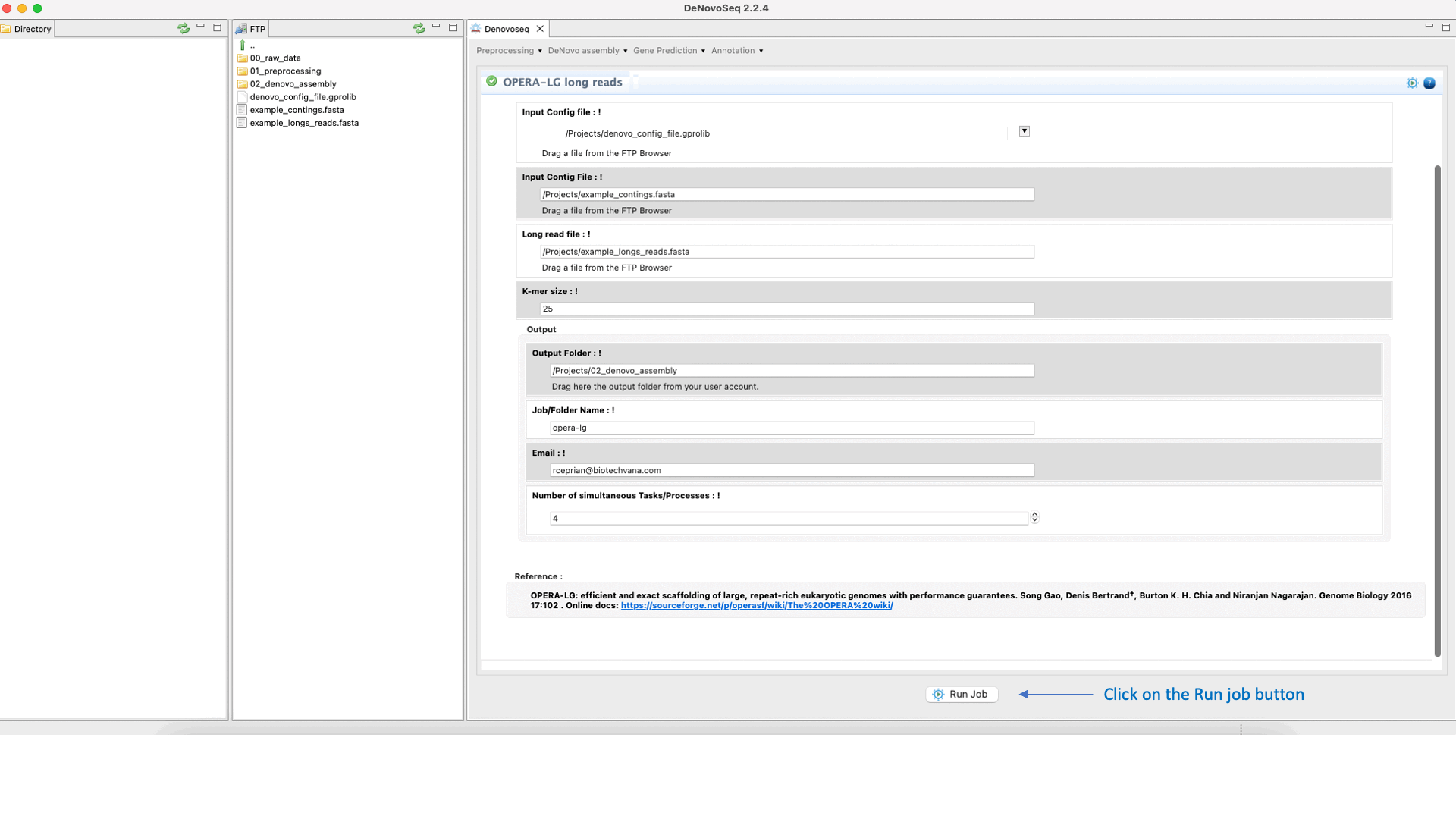
Figure 24: DeNovoseq interface to manage OPERA scaffolder.
Gene prediction refers to the set of methodologies and tools used to identify the genomic regions encoding for genes (protein-coding and non-coding) and other regulatory and functional elements. For prediction of prokaryotic genes, DeNovoSeq provides an ORF finder script also available in the SeqEditor application of the GPRO suite. For eukaryotic genomes, we provide an interface within DeNovoSeq to run AUGUSTUS (Stanke et al., 2008).
To run Find ORFs go to [Gene Prediction → Prokaryotes → Find ORFs]and follow Fig. 25.
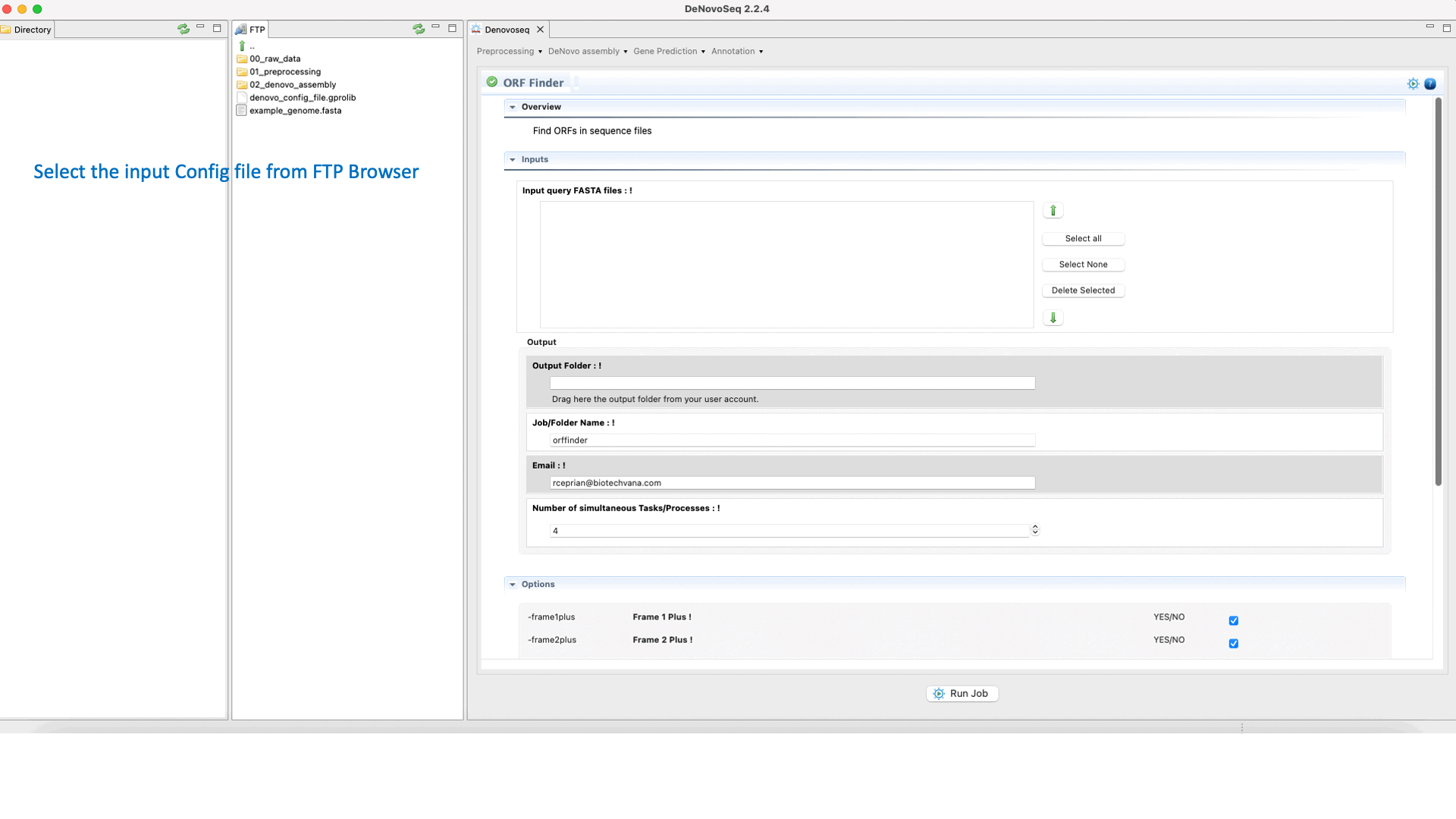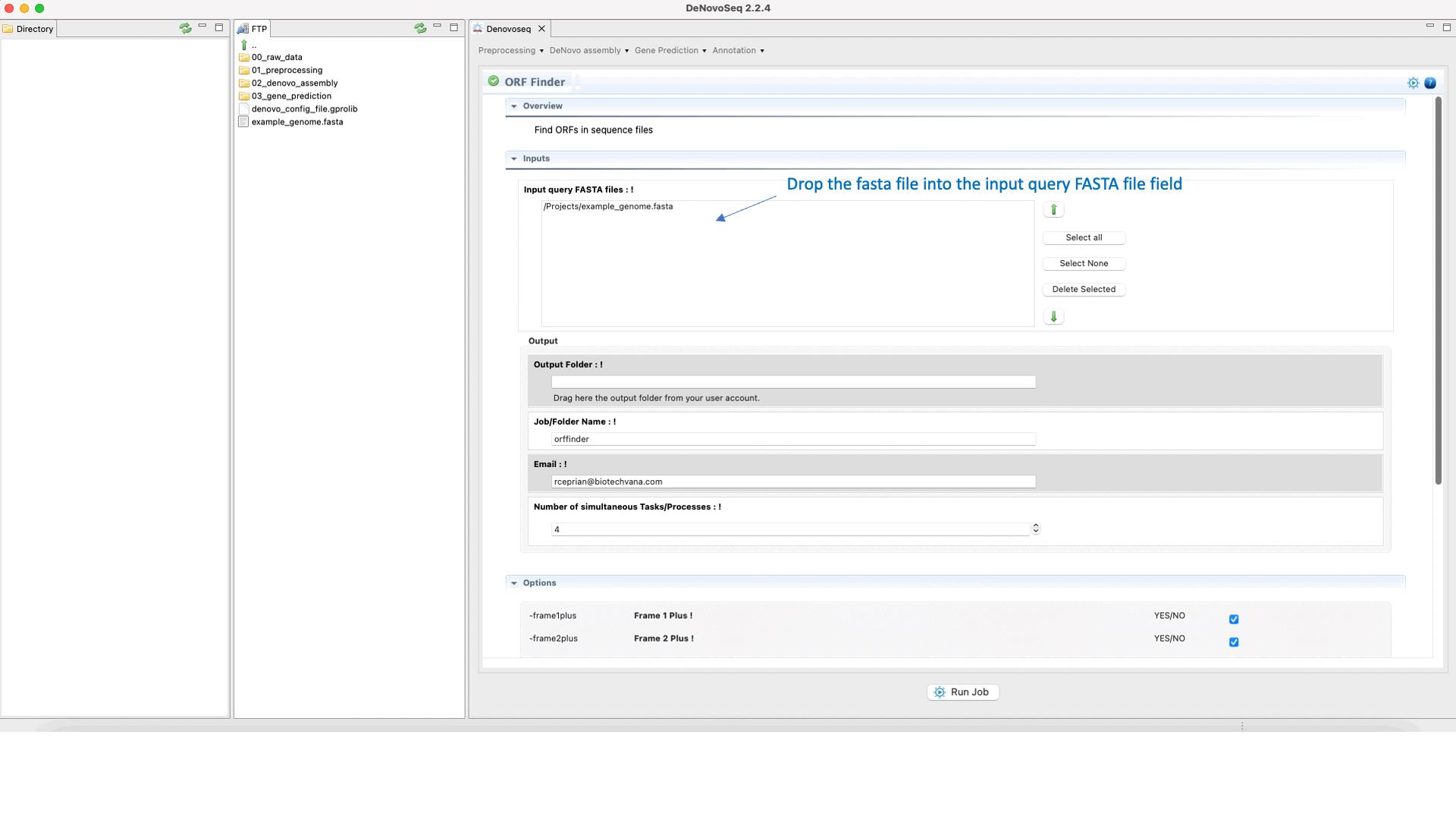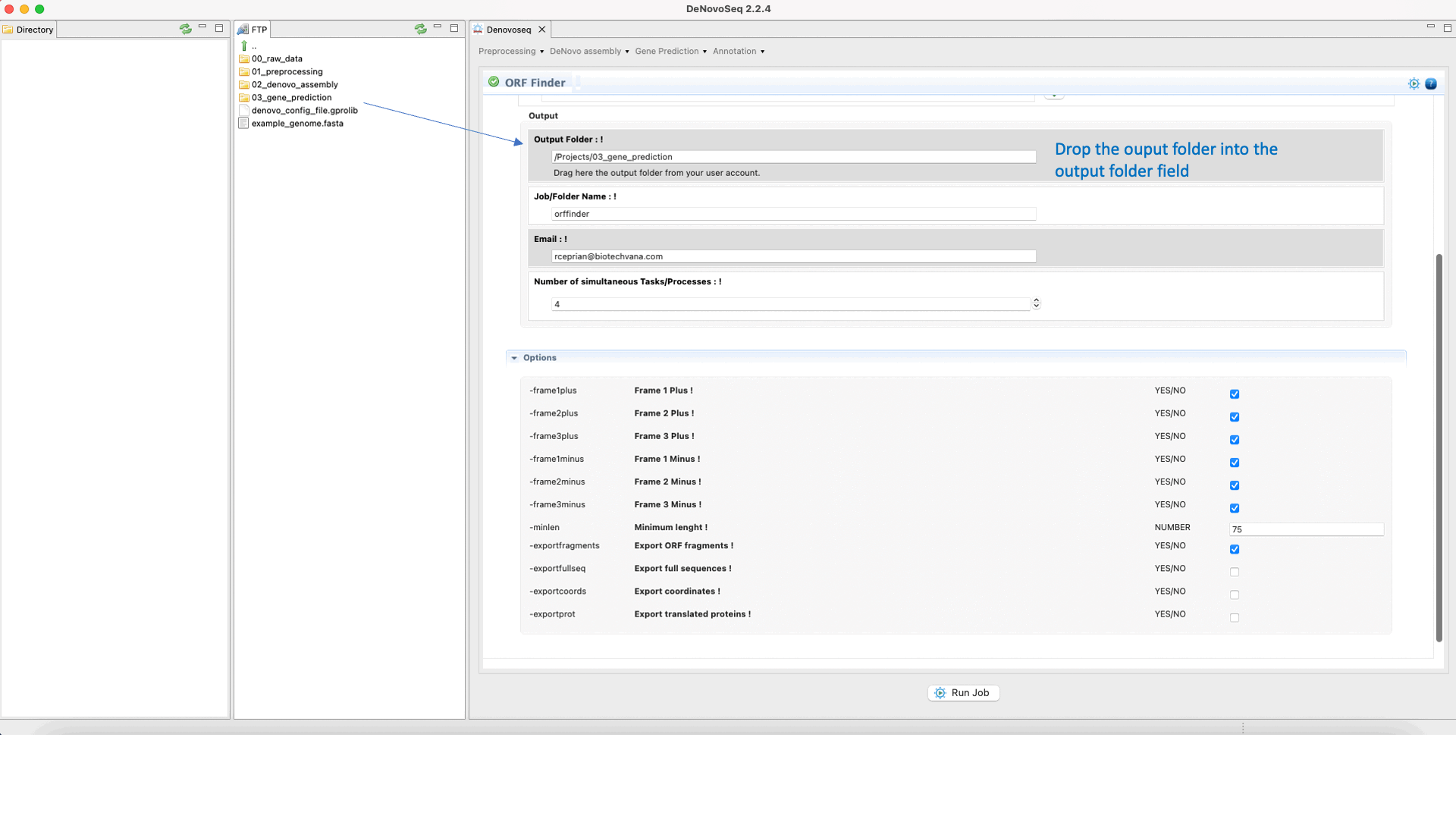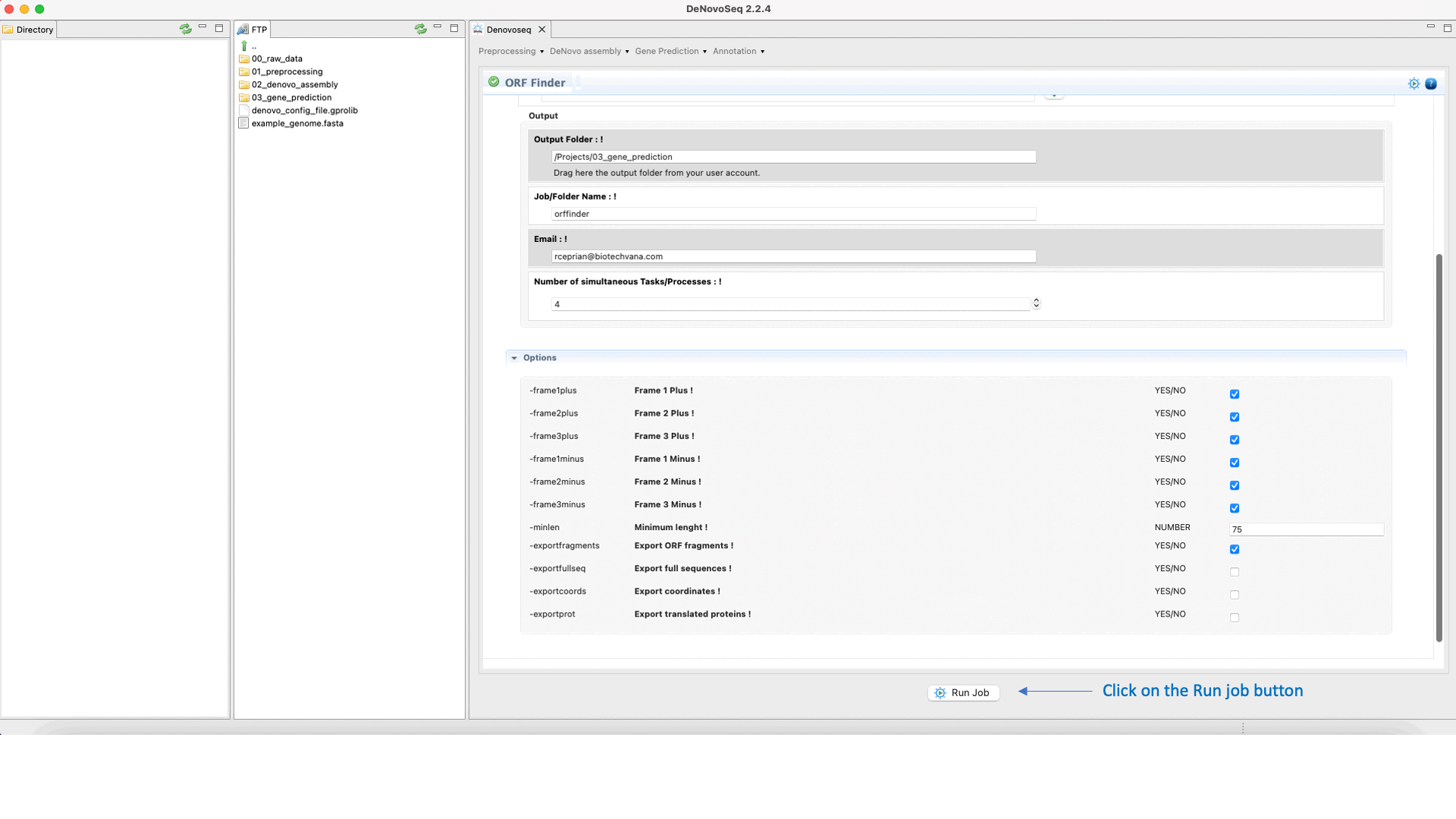
Figure 25: DeNovoSeq interface for using Find ORFs.
AUGUSTUS (Stanke et al., 2008) is an ab initio program that predicts genes from eukaryotic genome sequences based on a Generalized Hidden Markov probabilistic Model for a sequence and its gene intron-exon structure. The implementation of AUGUSTUS in DeNovoSeq is a workflow that contemplates three steps; “Training”, “Hints preparation” and “Prediction”. See the AUGUSTUS manual at "http://bioinf.uni-greifswald.de/augustus/" for more information
.To run the Training step of AUGUSTUS go to [Gene prediction → Augustus → Training]and follow Fig. 26.
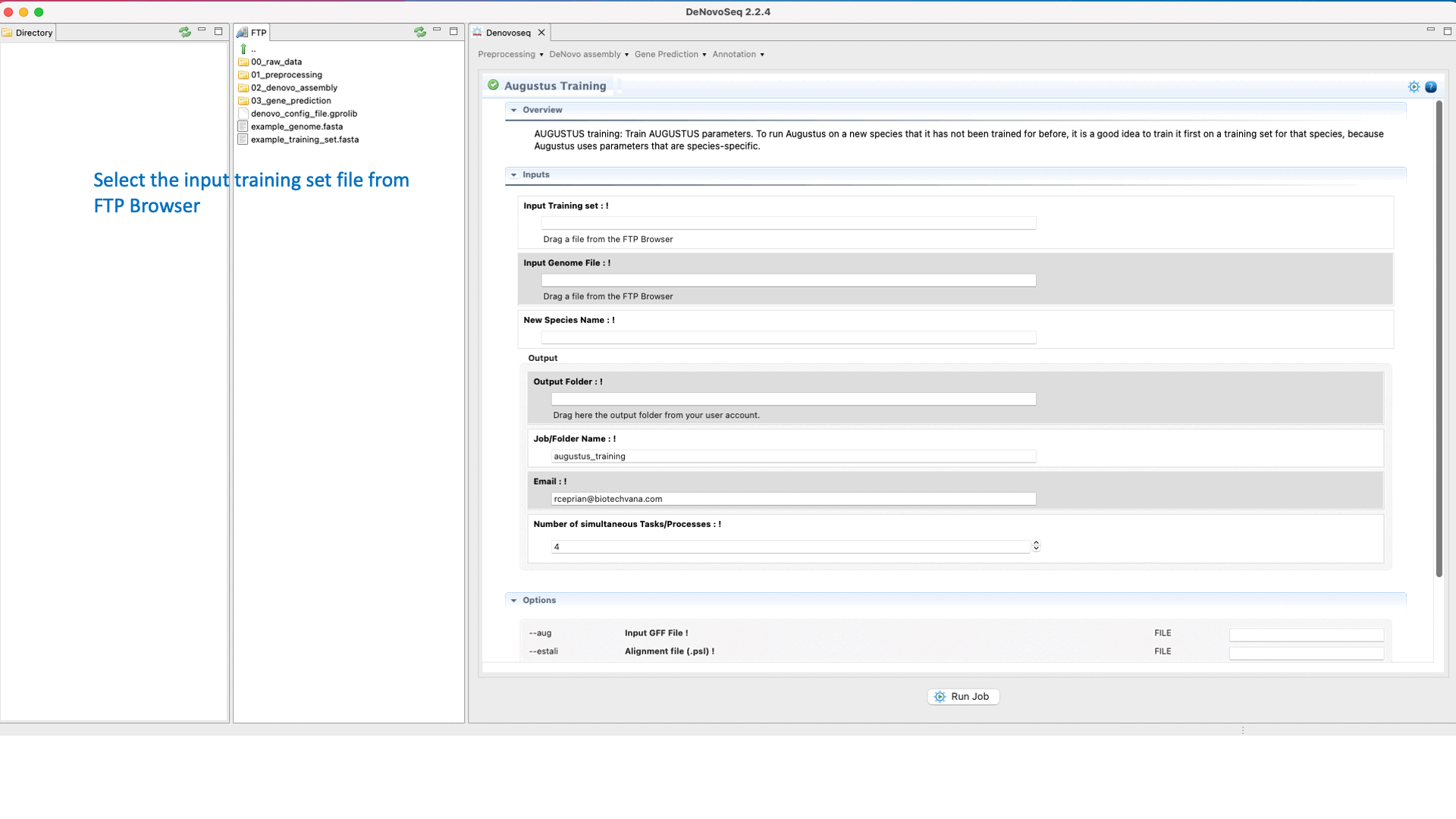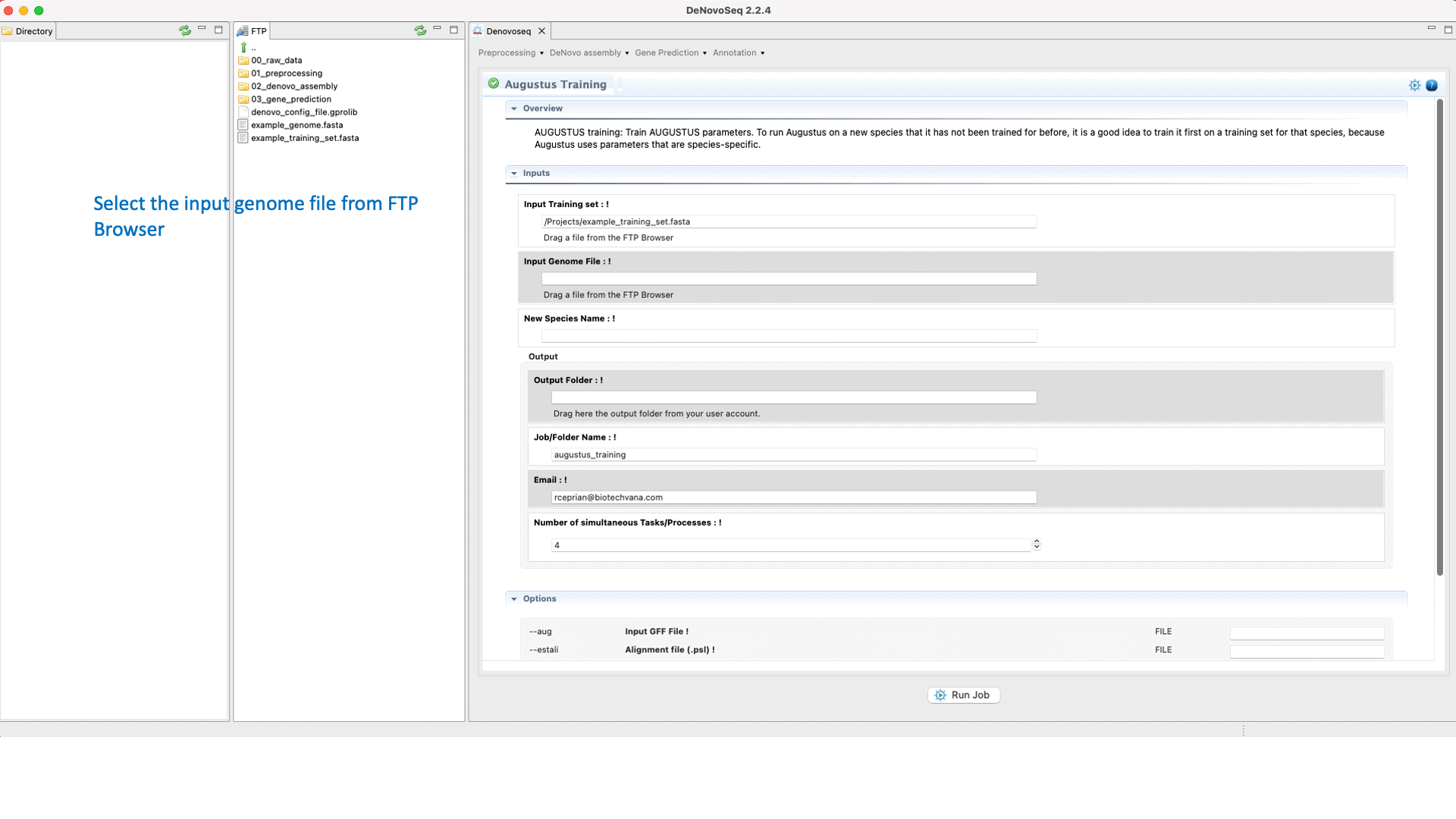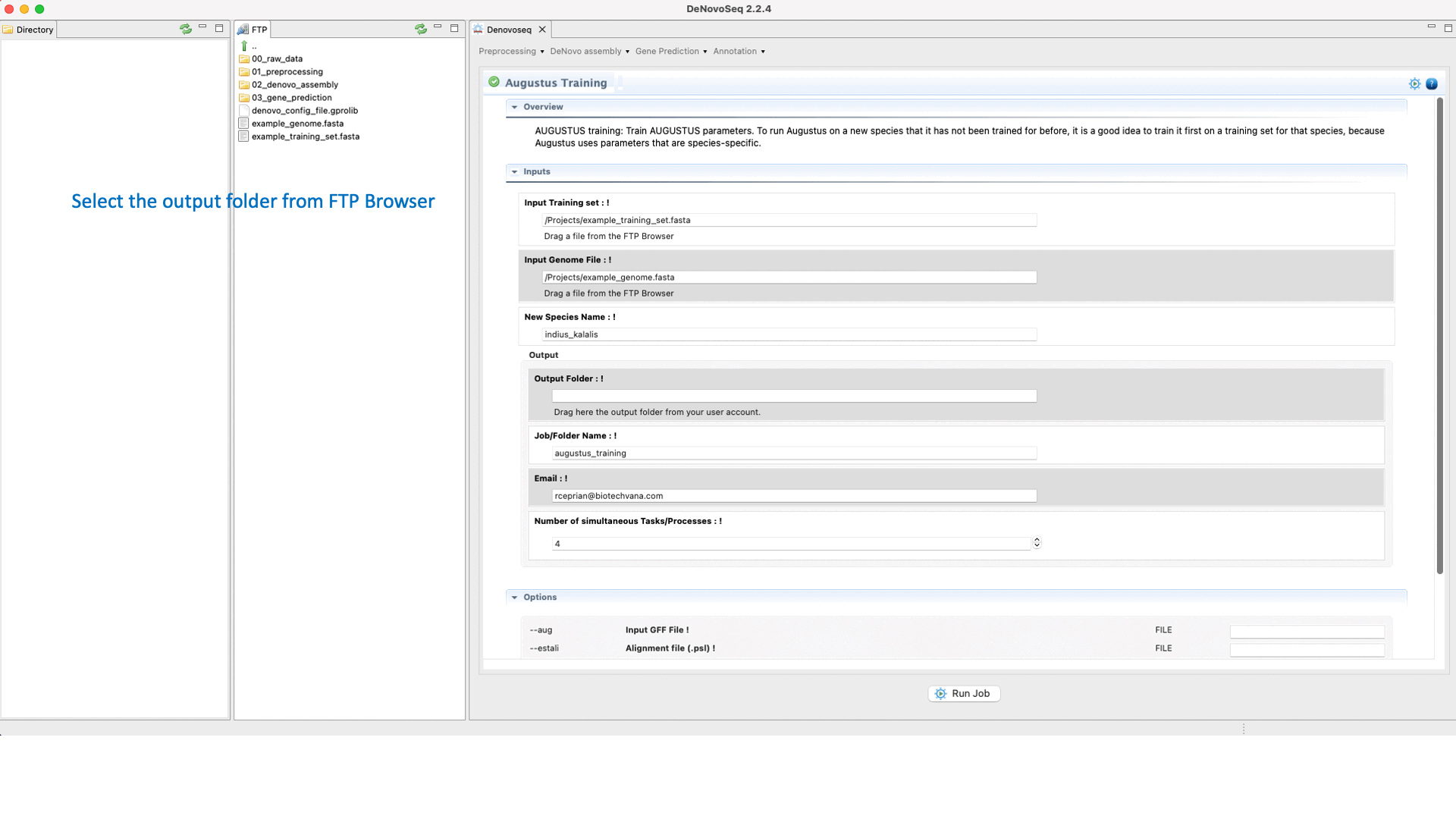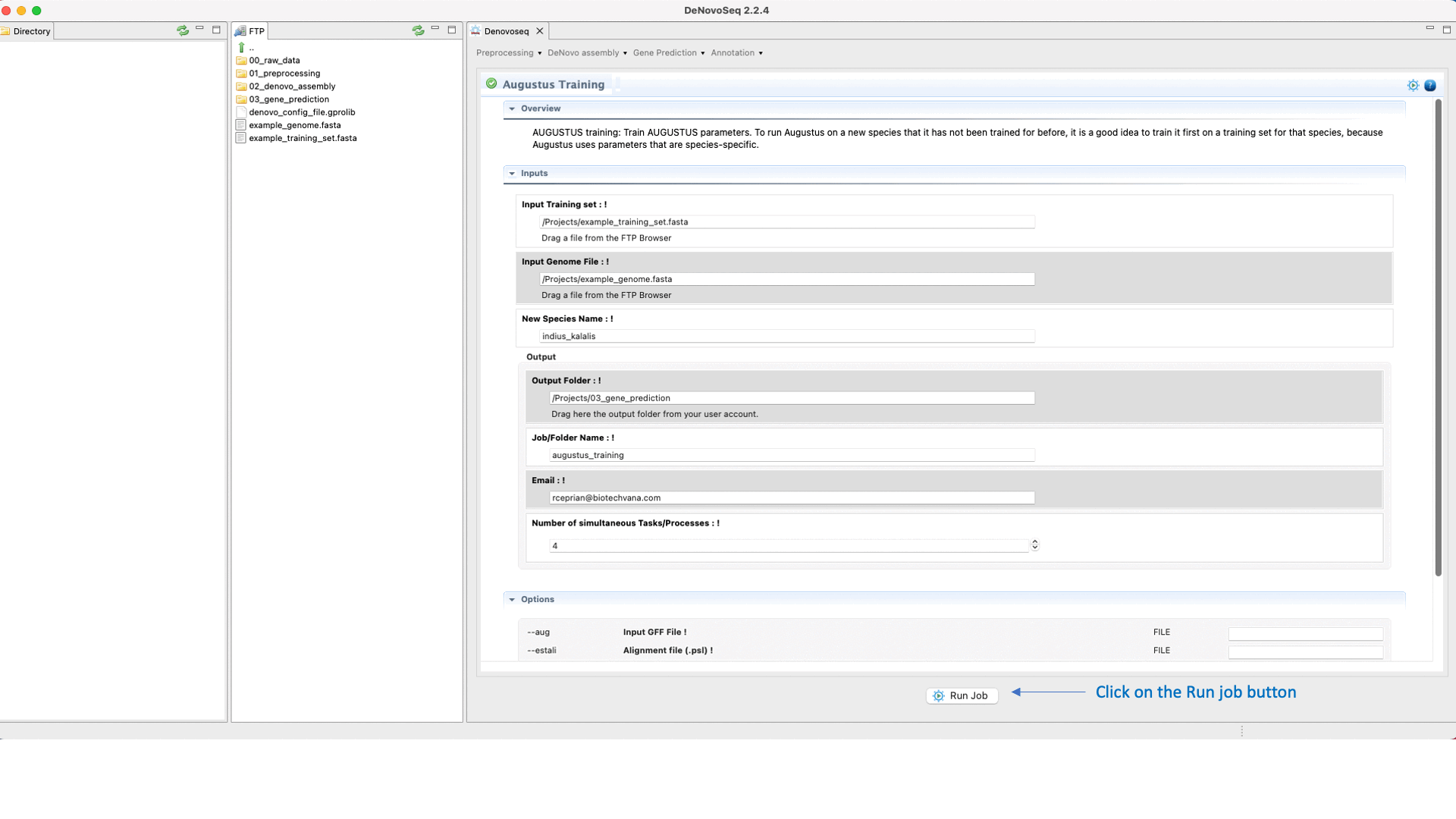
Figure 26: DeNovoSeq interface to train AUGUSTUS for predicting gene structures in your genome.
Users can also incorporate hints on the gene structure from extrinsic sources to improve the accuracy of the gene prediction. Some examples of allowed hints follow:
If you have the needed material to create the hints then go to [Gene prediction → Augustus → Hints]and follow Fig. 27.
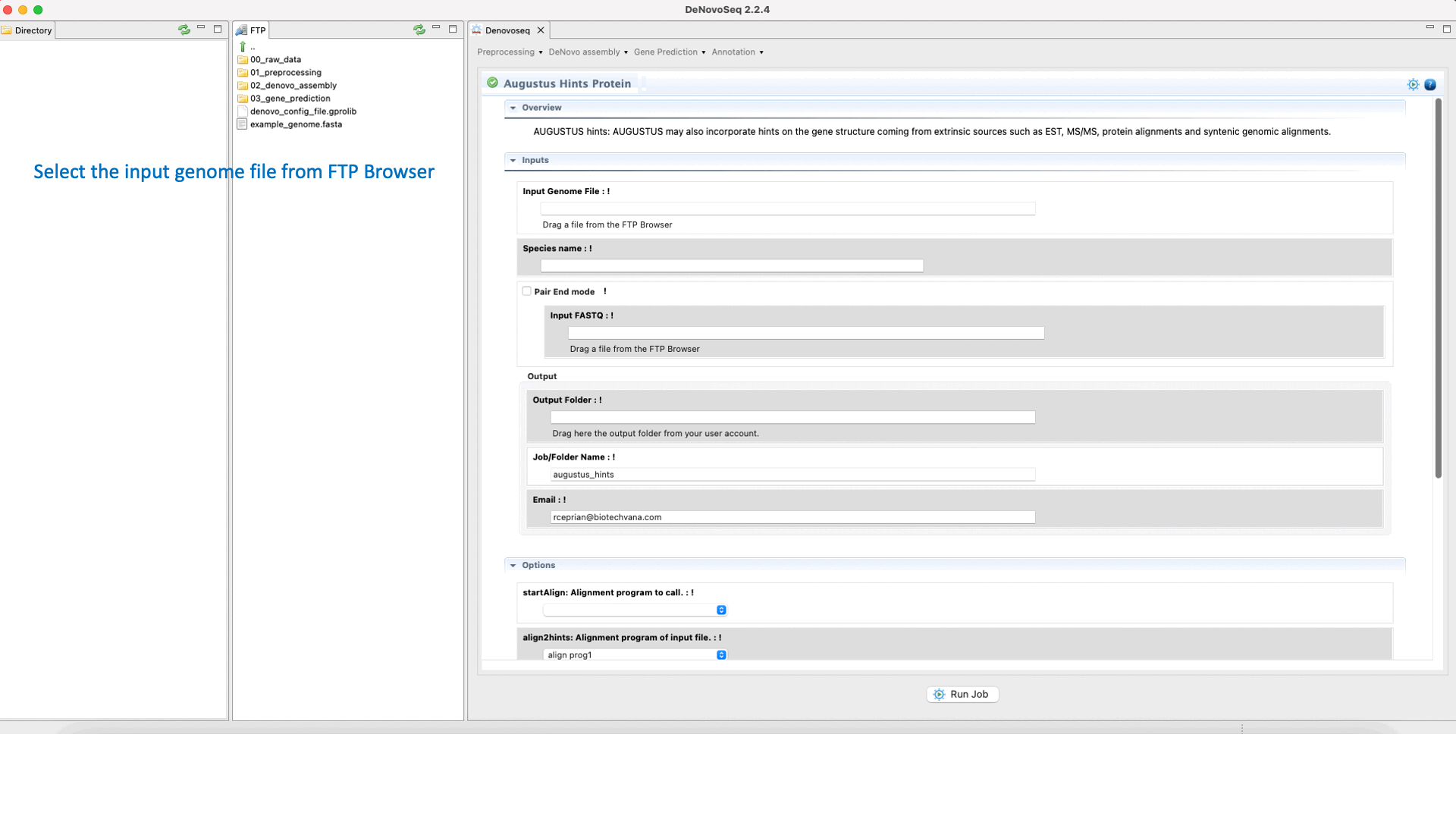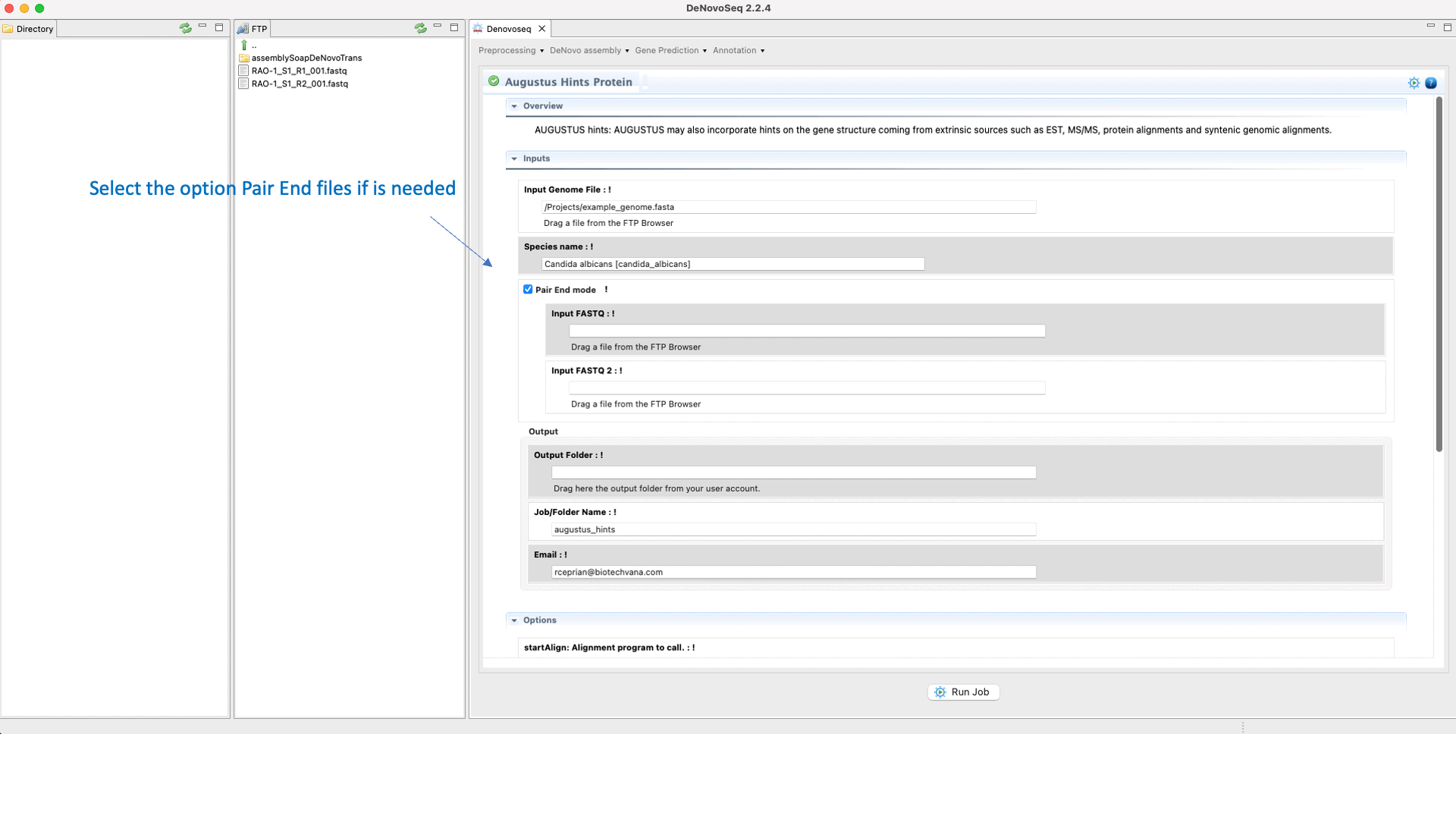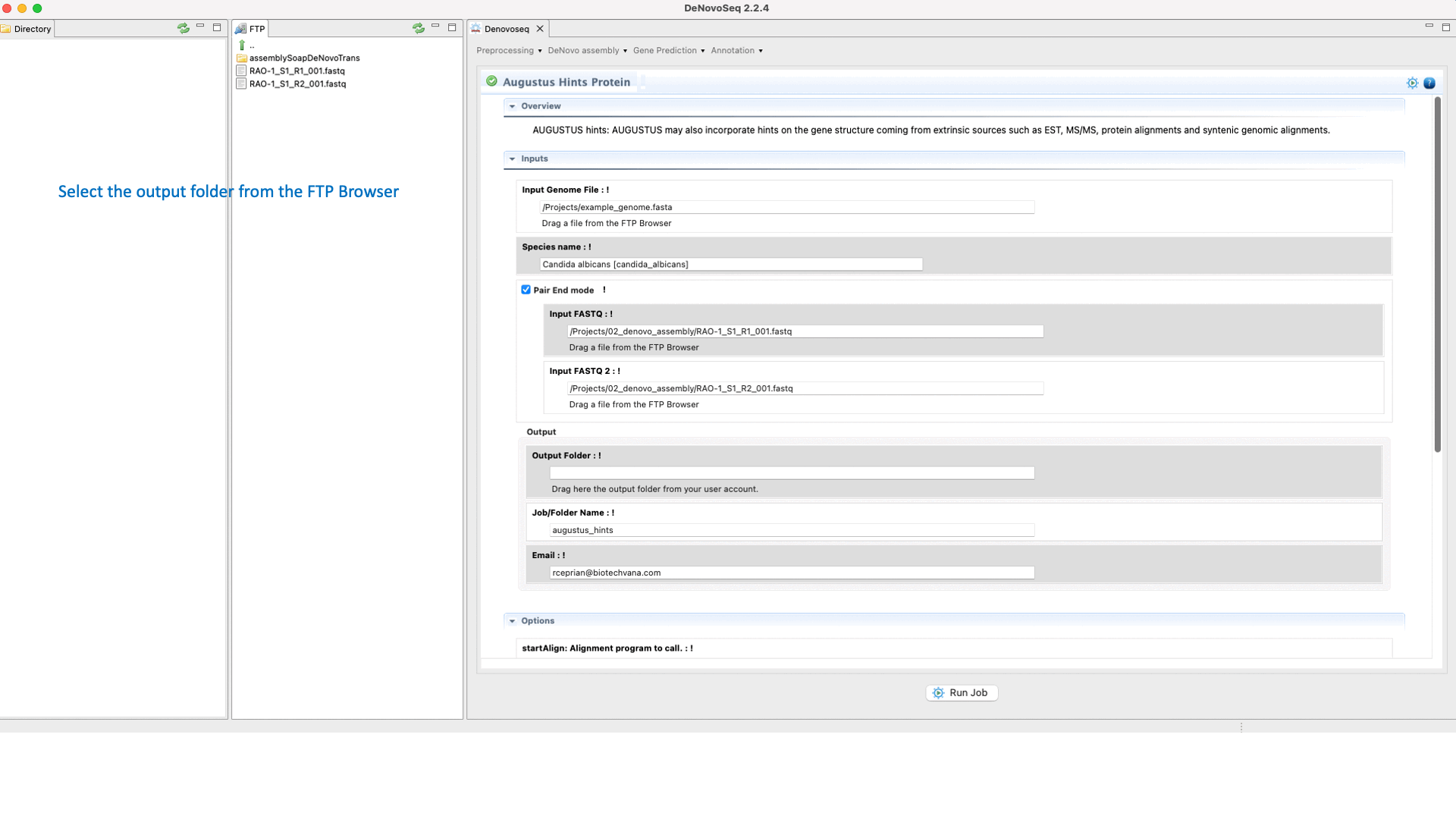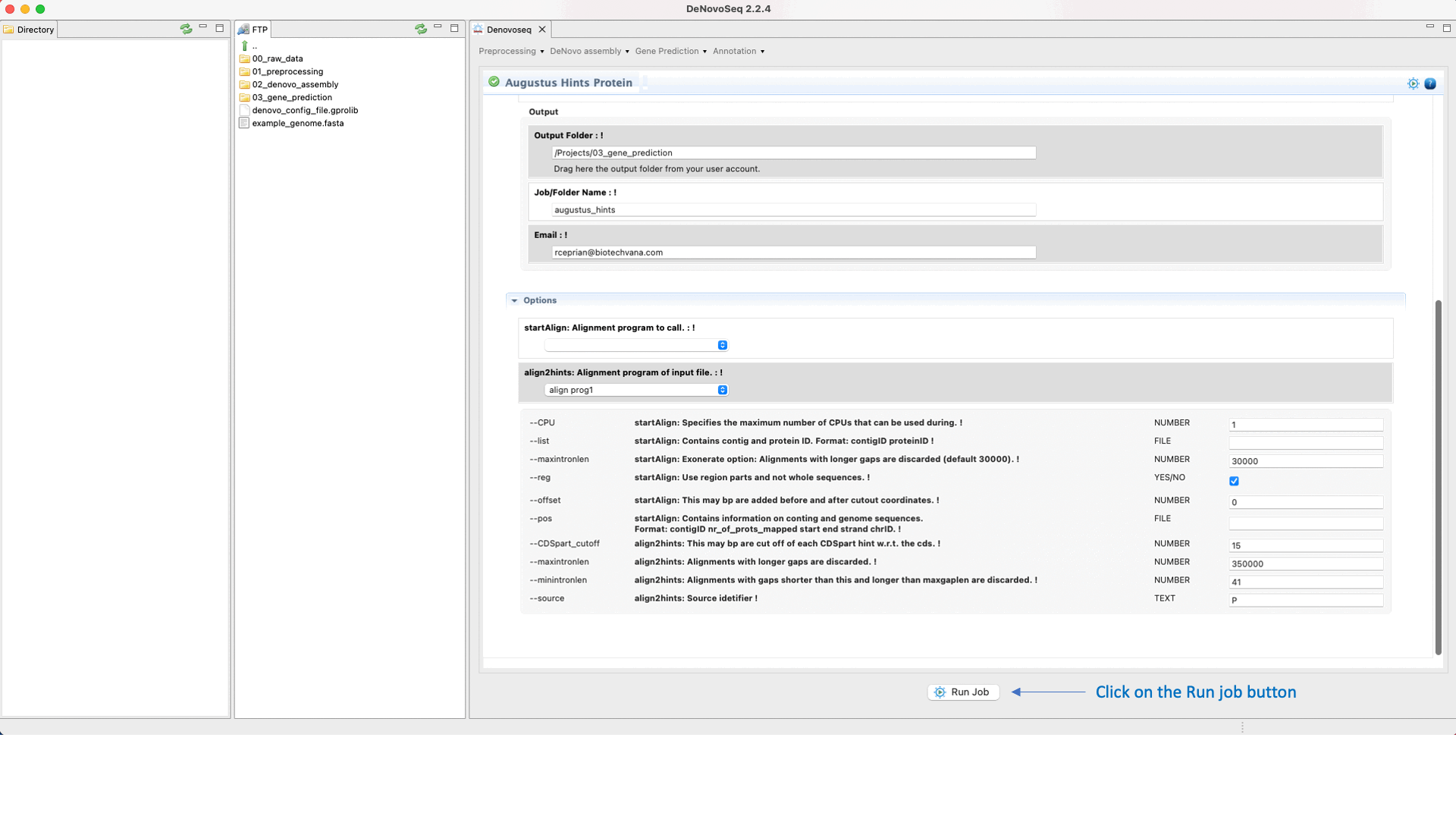
Figure 27: DeNovoSeq interface to create the hints with AUGUSTUS
Once the training step has been completed and the hints (optional) have also been created, users can run the Predictions step going to [Gene prediction → Augustus → Prediction]and follow Fig. 28.
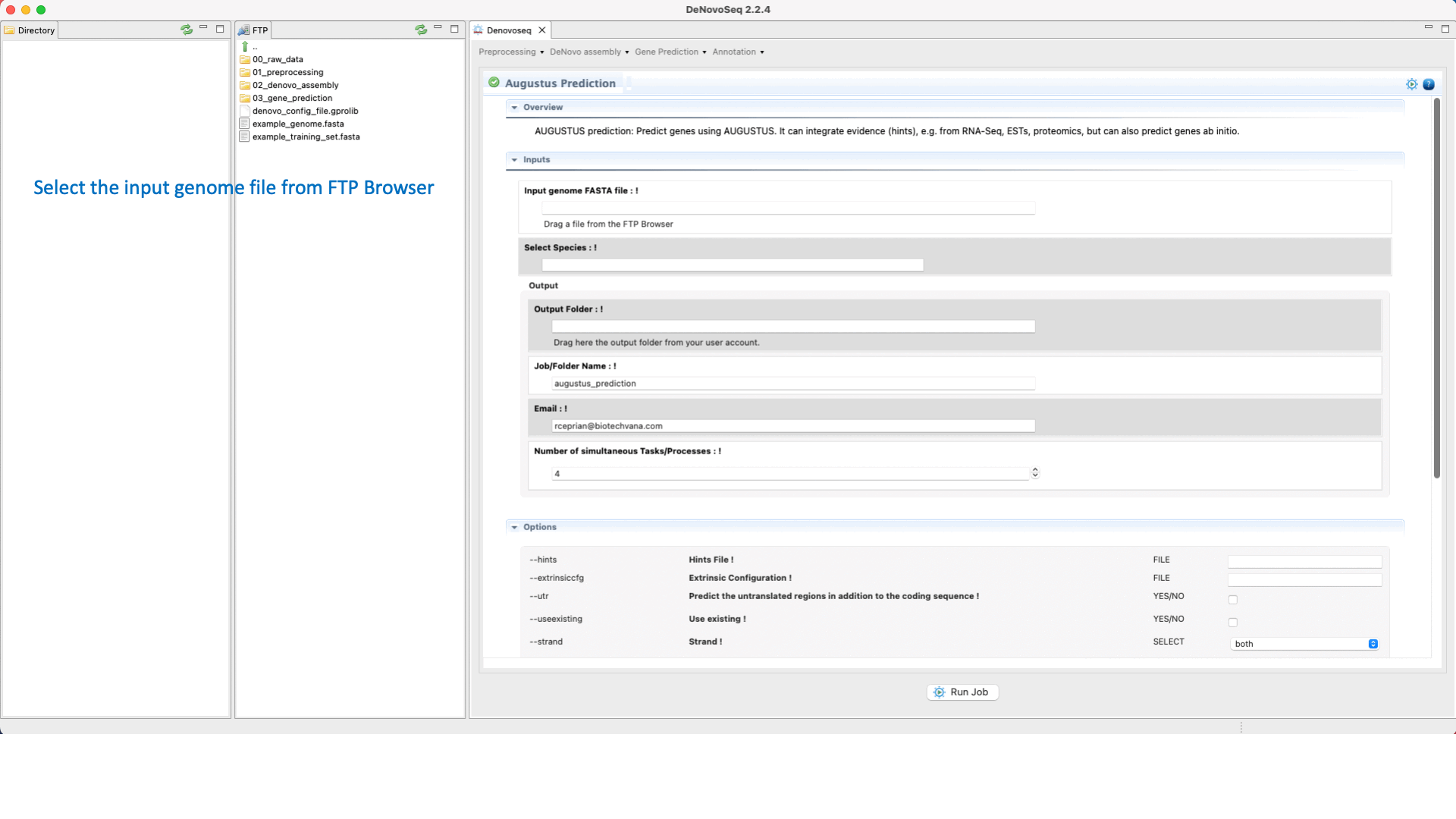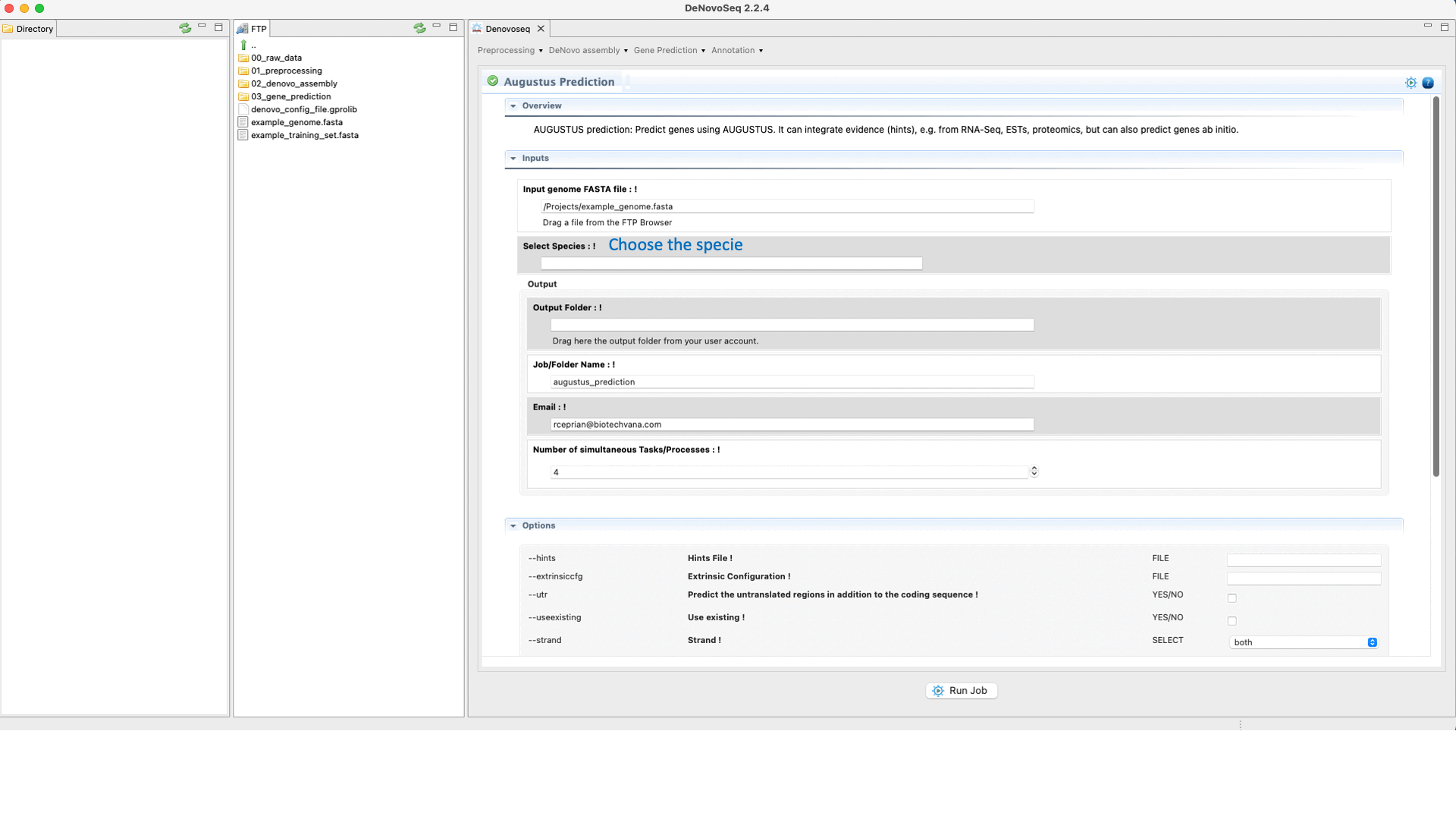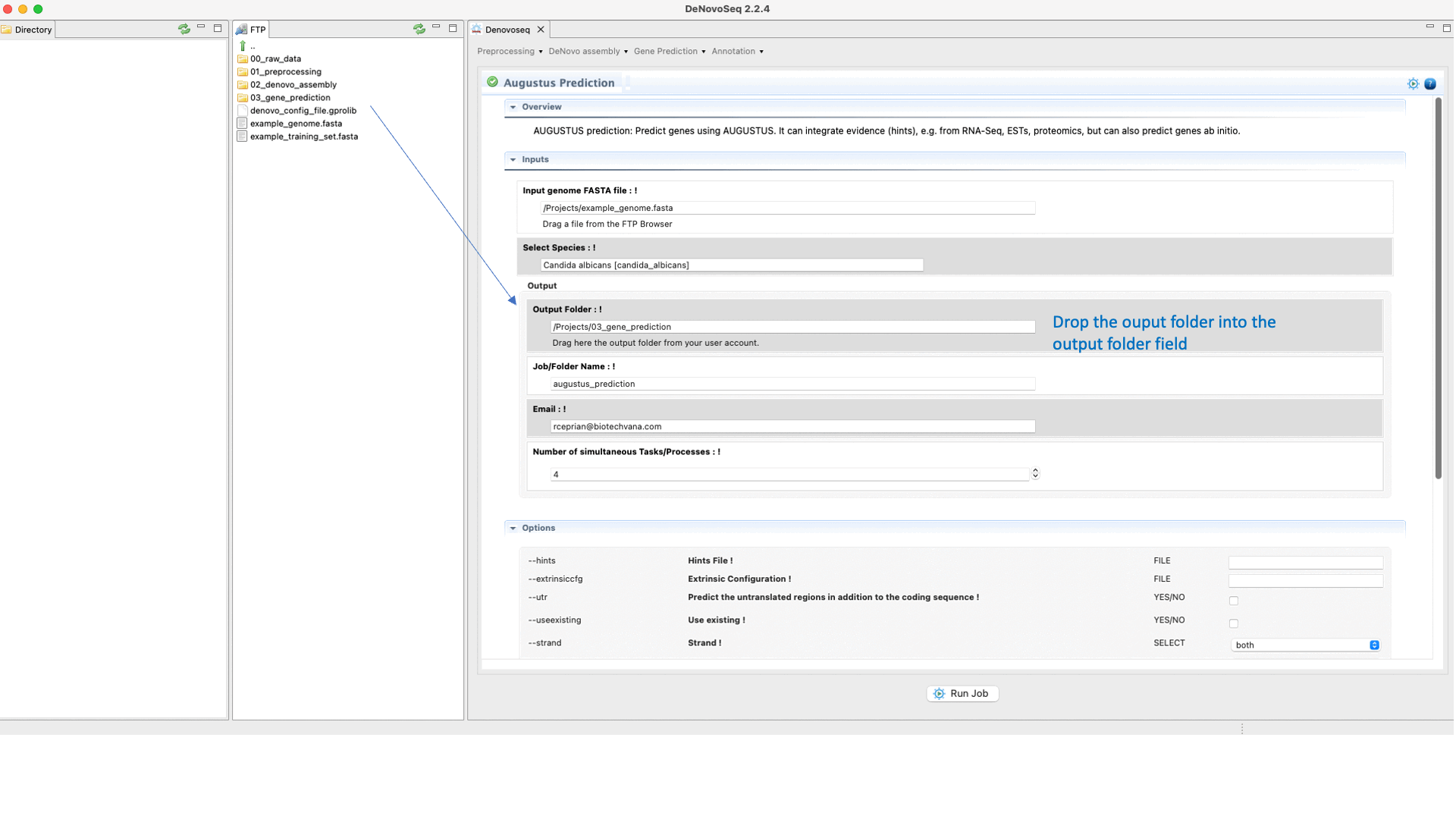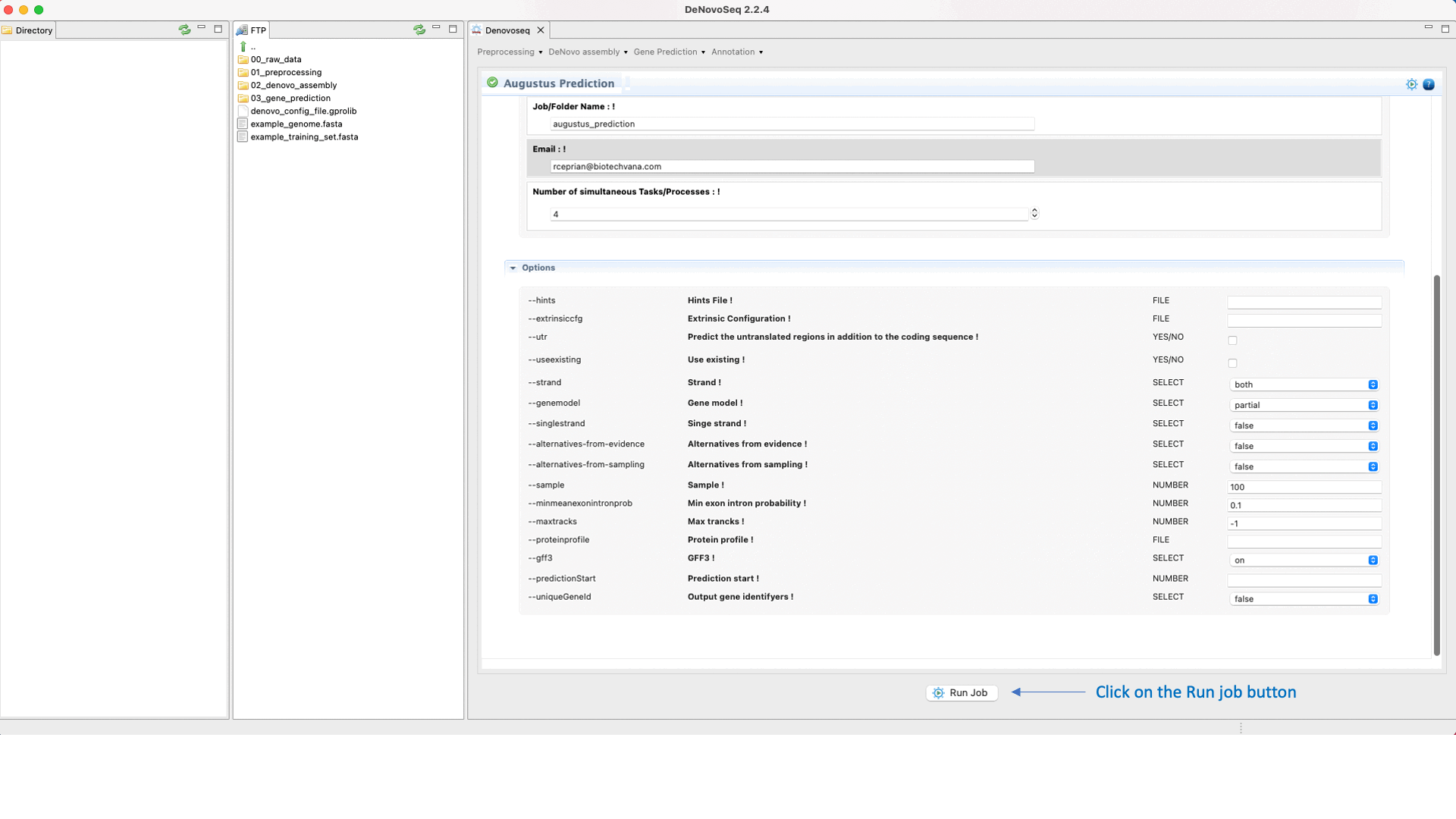
Figure 28: DeNovoSeq interface to perform the Gene prediction using AUGUSTUS.
In deNovo protocols, annotation is the step (normally final) oriented to identify function, domains and biological roles of a set of predicted genes and transcripts (coding and not coding). The annotation is normally performed via sequence-to-sequence or sequence-to-profile alignment comparison to find statistically significant homologies between your query sequences and a RefSeq database. DeNovoSeq permits the user to perform the annotation either by manual or by automatic means using three of the most tools for automatic annotation; NCBI BLAST package Altschul et al., 1990, HHMER3 Mistry et al 2013
.NCBI BLASTAltschul et al., 1990is a software package that finds regions of local similarity between a query sequence (or a file in fasta format with the query sequences) and the subjected sequence models or refseq database searched by the query/s. The package implements different tools to compare nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches:
For more information see the NCBI-BLAST manual at "https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs".
The step-by-step mode of DeNovoSeq presents a specific interface for each step of a typical BLAST analysis organized in the Annotation tab, as follows:
- Format BLAST databases
To perform a BLAST analysis the subject database must be formatted in BLAST format. For this task, DeNovoSeq implements an interface that accepts both protein and nucleotide fasta files as input formatting them for BLAST.
To do this go to [Annotation → NCBI-BLAST → Format databases] and follows what is stated in Fig.29 for formatting blast subjects.
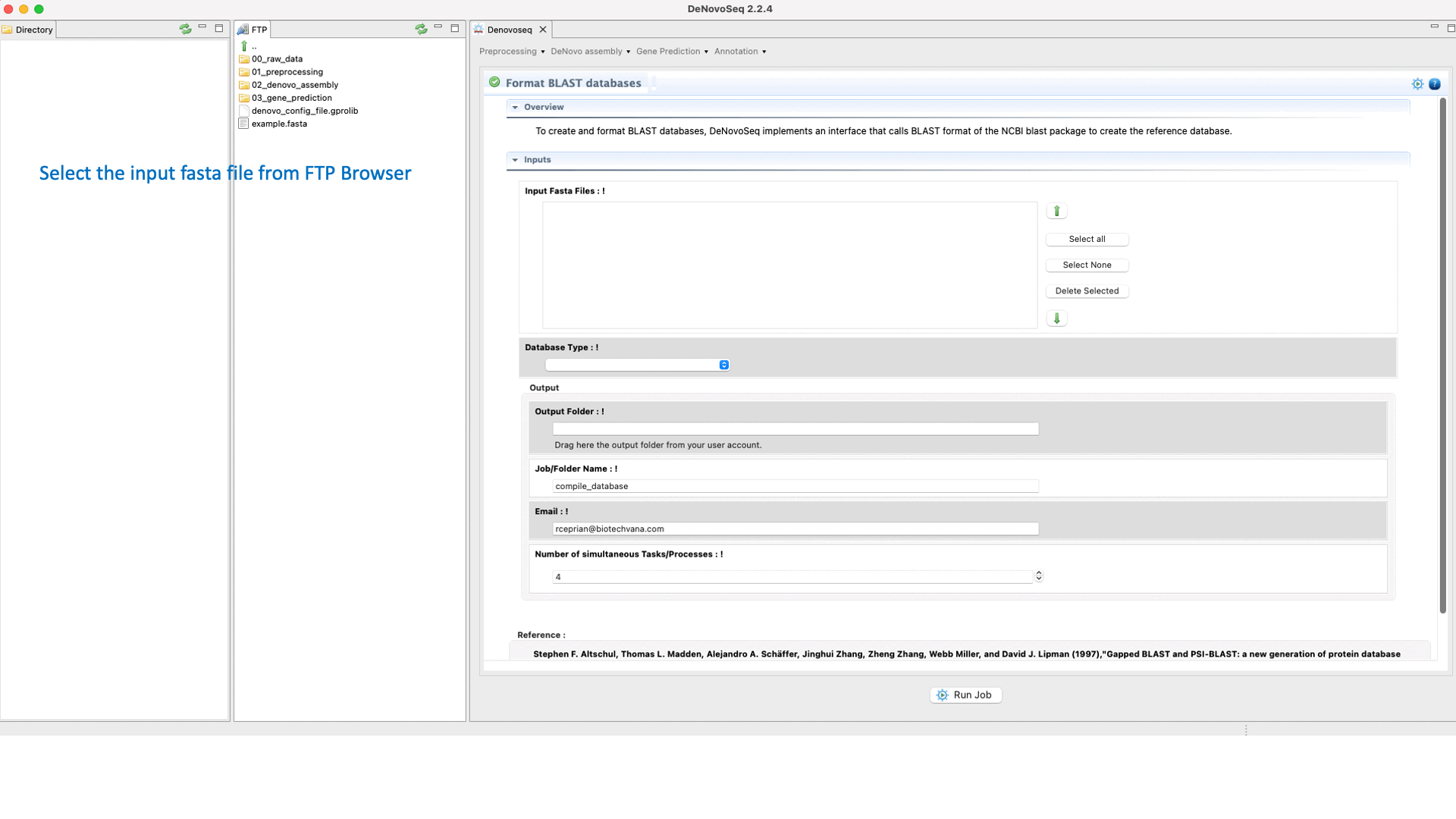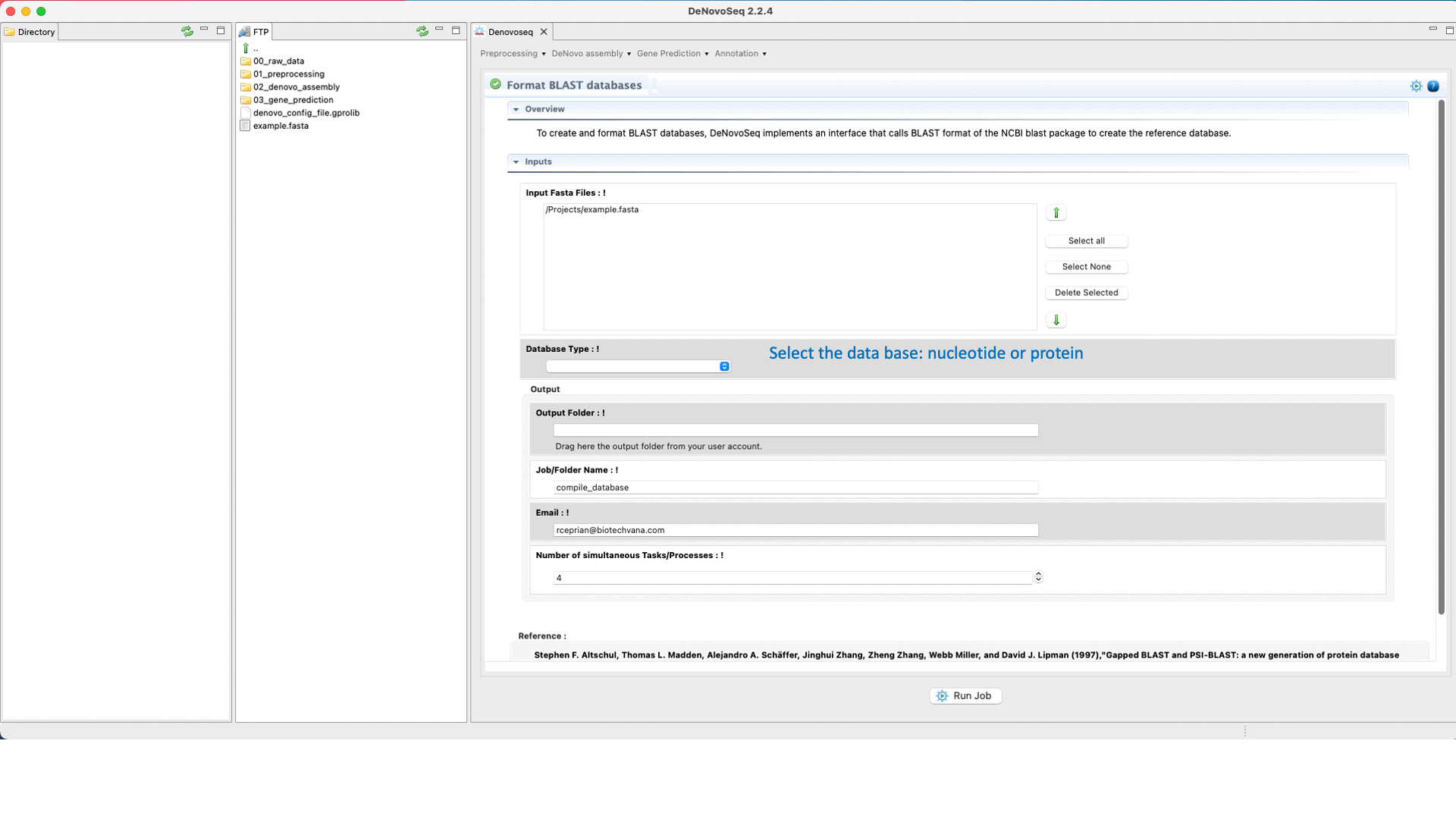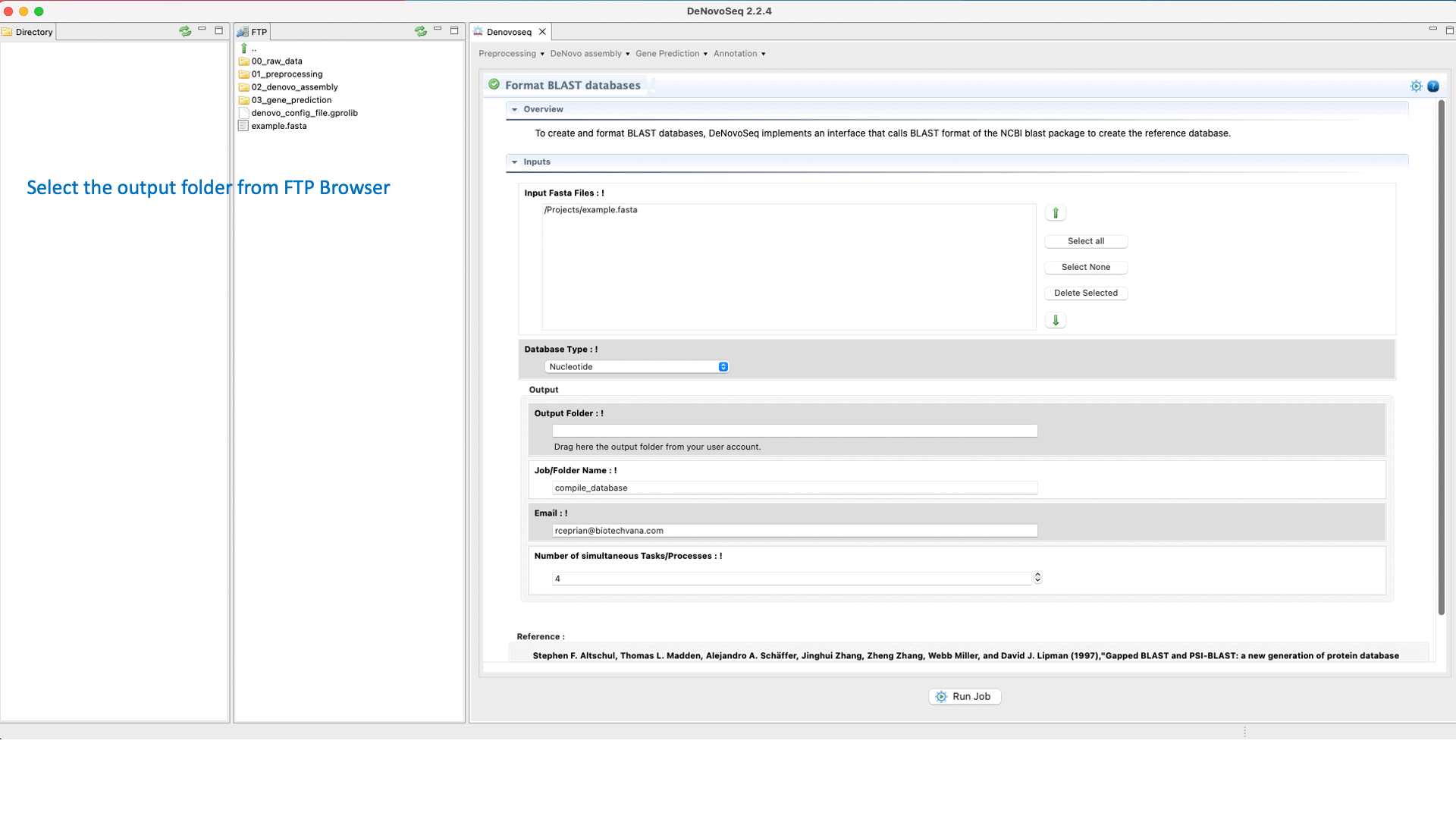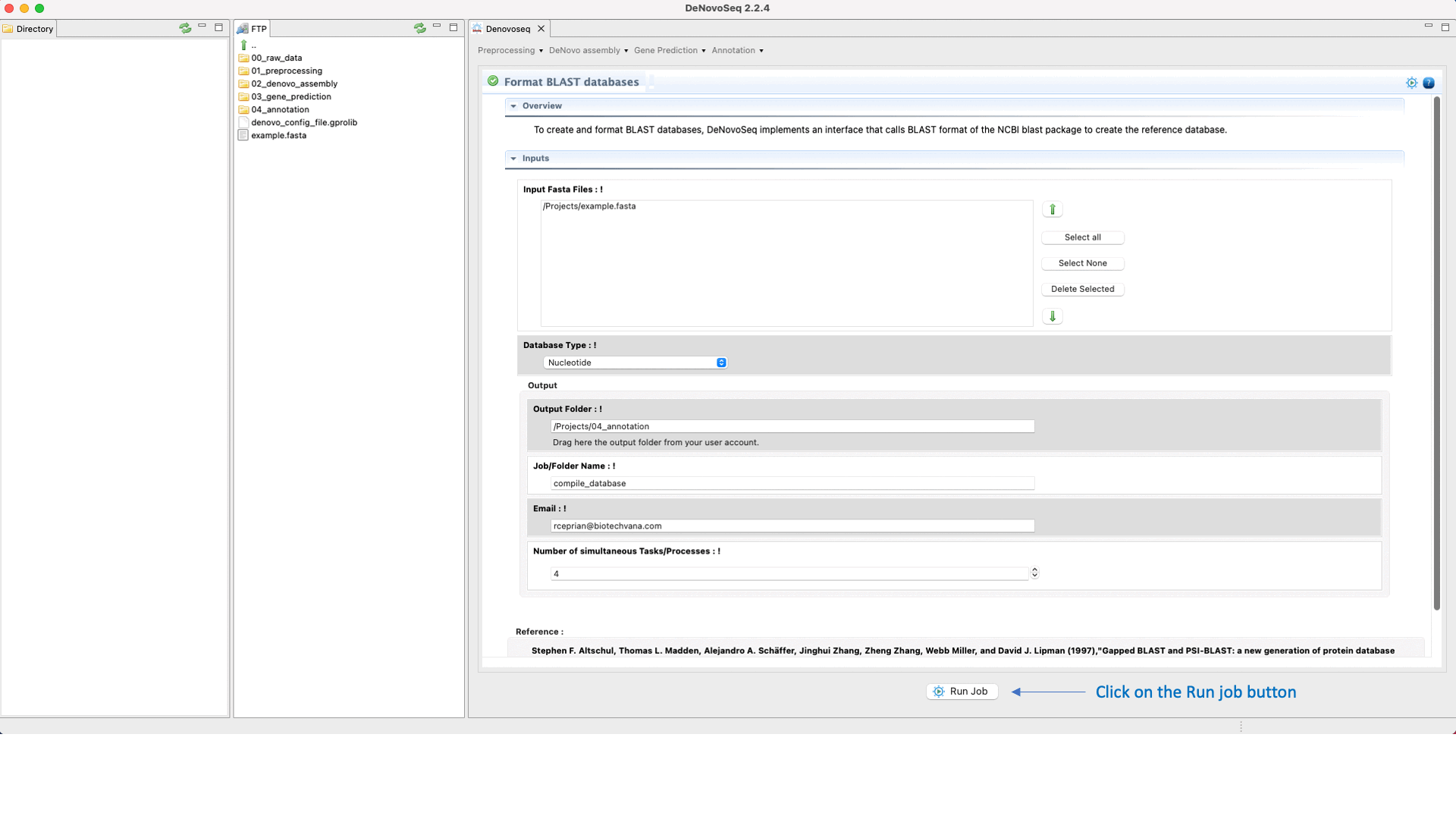
Figure 29: Using the GPRO interface for formatting blast subjects.
- Import RefSeq databases
Big databases such as those provided by NCBI (NR, RefSeq) or Uniprot (Swissprot) are very difficult to process and occupy a significant part of the disk size in your PC or in your user account if you are working on the server side.
For users working on the server side, we facilitate a centralized repository for big databases whose links can be imported to user account going to [Annotation → NCBI-BLAST → Import BLAST databases] and doing as indicated in Fig.30 for importing RefSeq databases.
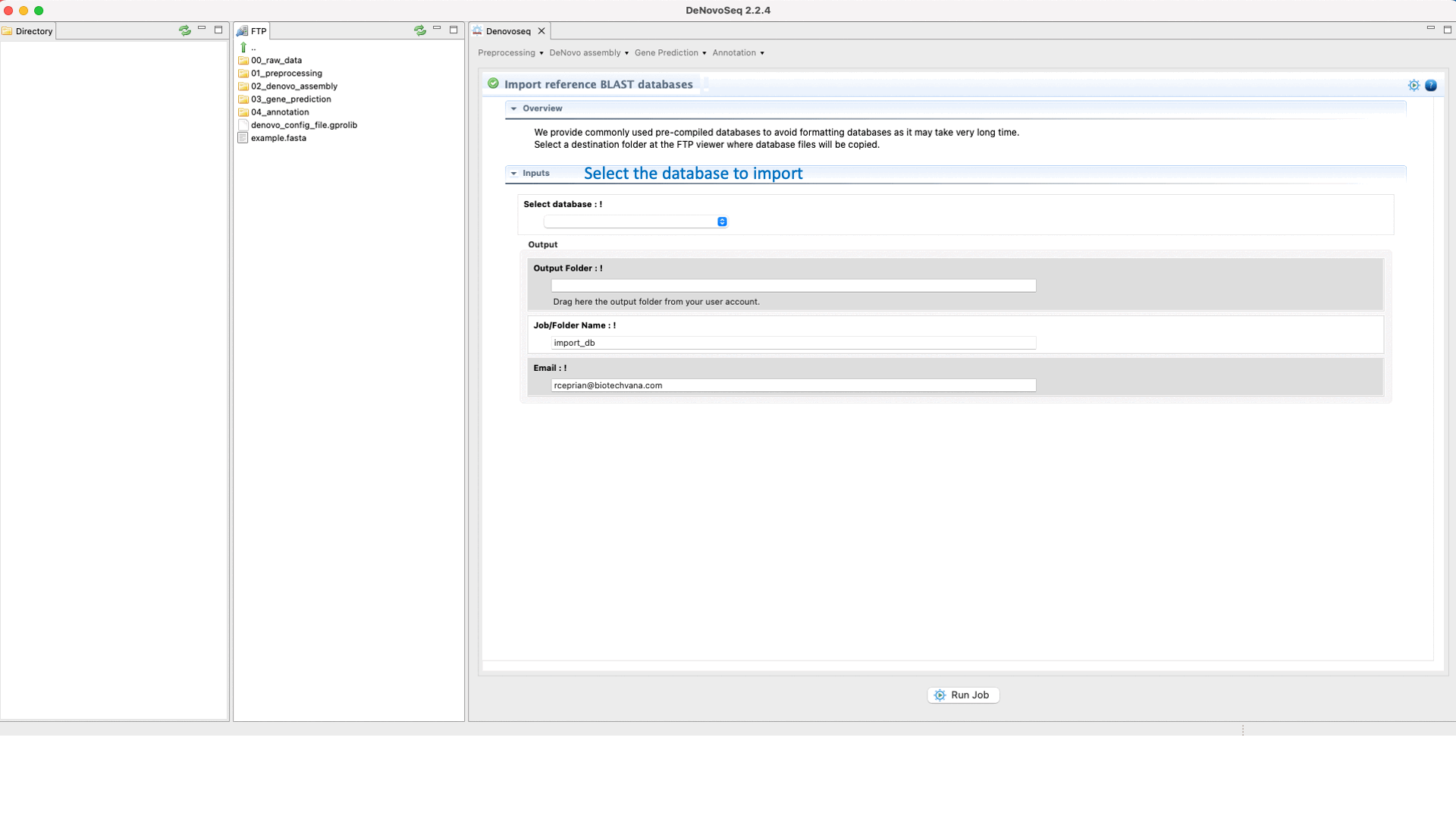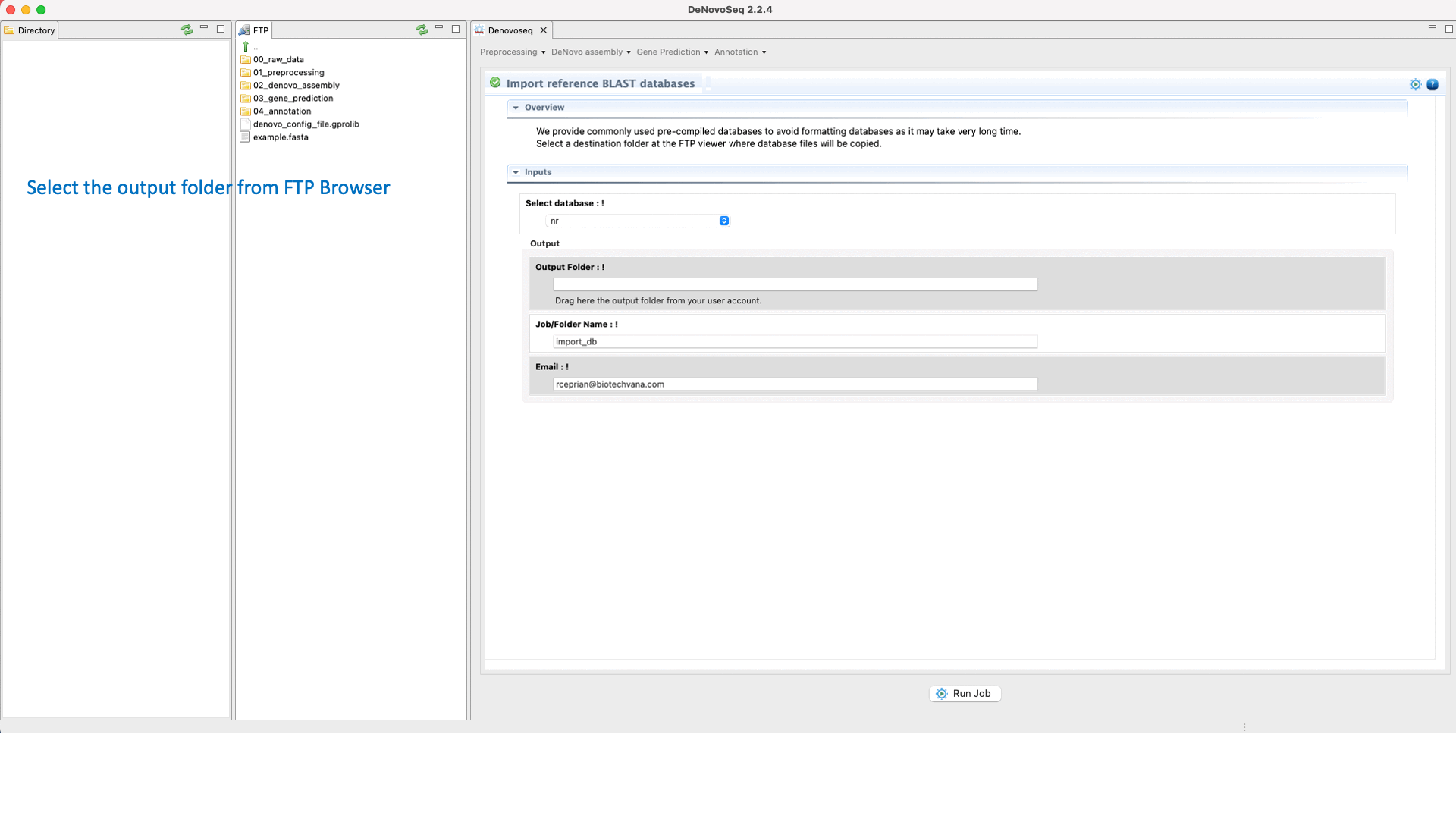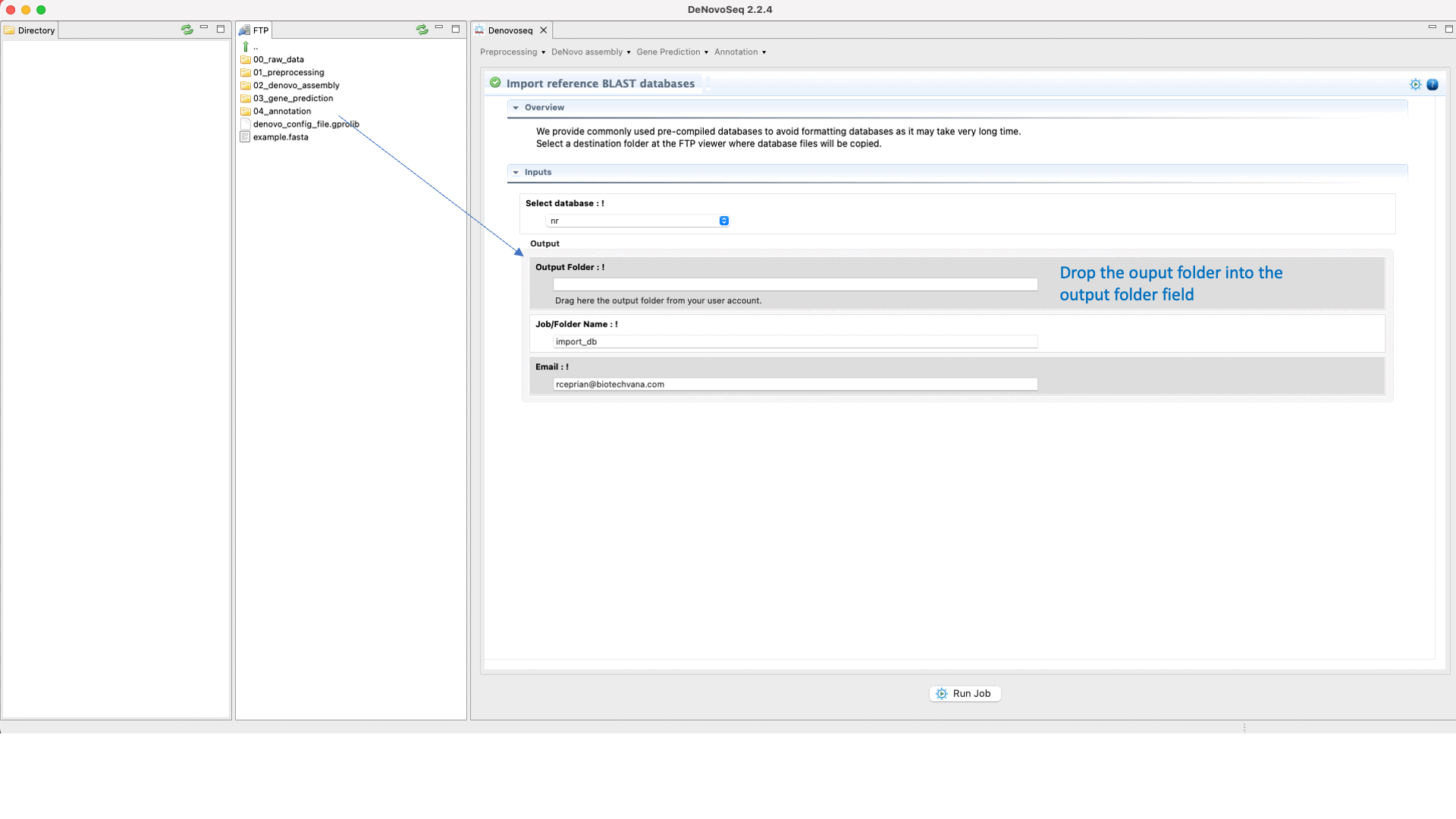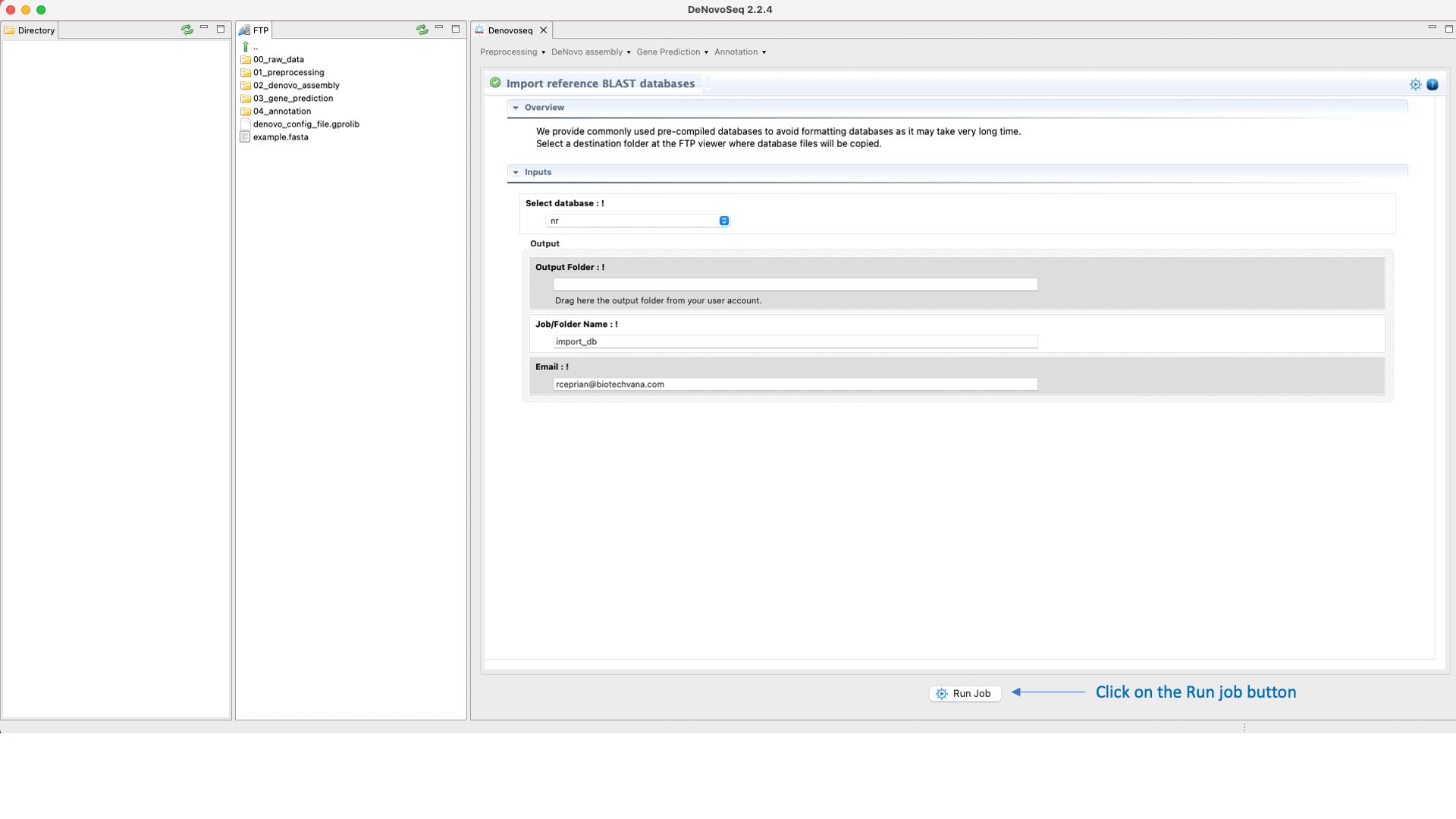
Figure 30: Using the GPRO interface to format fasta files as NCBI-BLAST reference databases or for importing them precompiled.
- BLAST search with fasta file query
DeNovo Seq provides a frame for the conventional web-based interface of the NCBI-BLAST allowing the users to make fast searches against their databases and multifasta files using one or multiples sequences queries and obtaining the typical alignment output provided by BLAST . Annotation of multifasta files with multiple sequences (for example genomes, transcriptomes o proteomes) may take a significant time to complete the process and deliver the results (hours or perhaps days depending on the number of sequences per file).
To execute the BLAST search for single queries go to [Annotation → NCBI-BLAST → BLAST search with one query] and follows what stated for single queries in Fig.31.
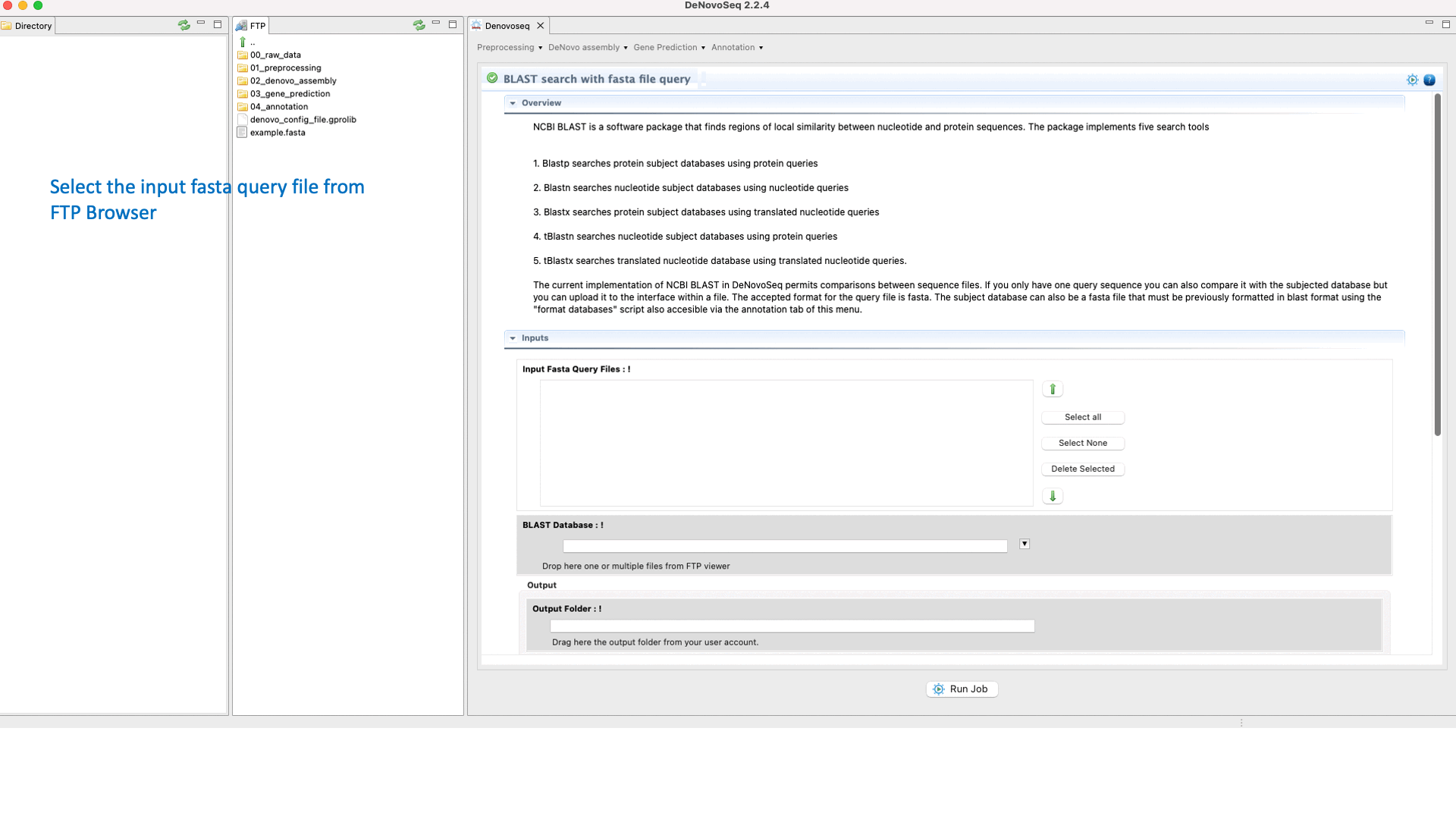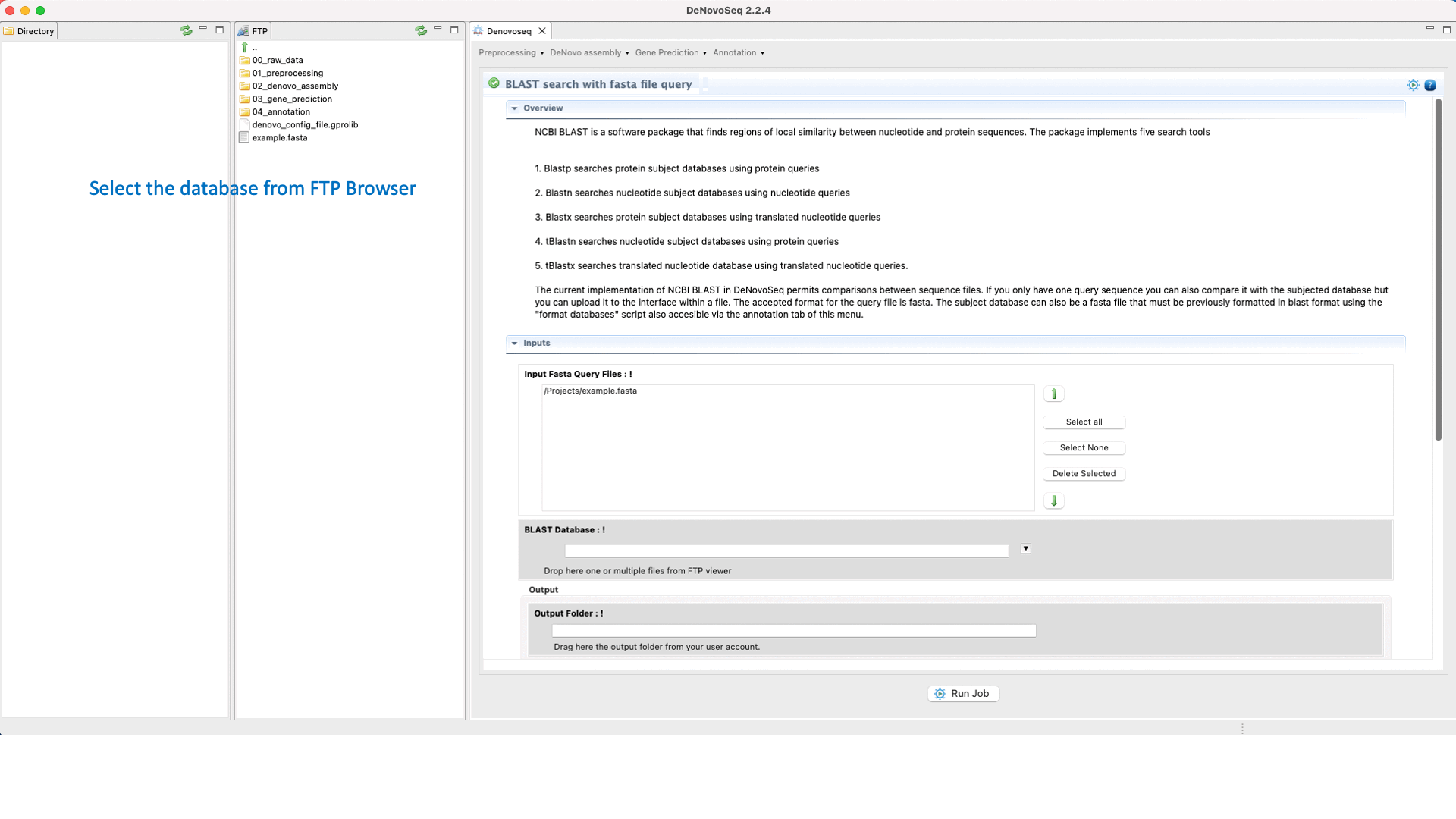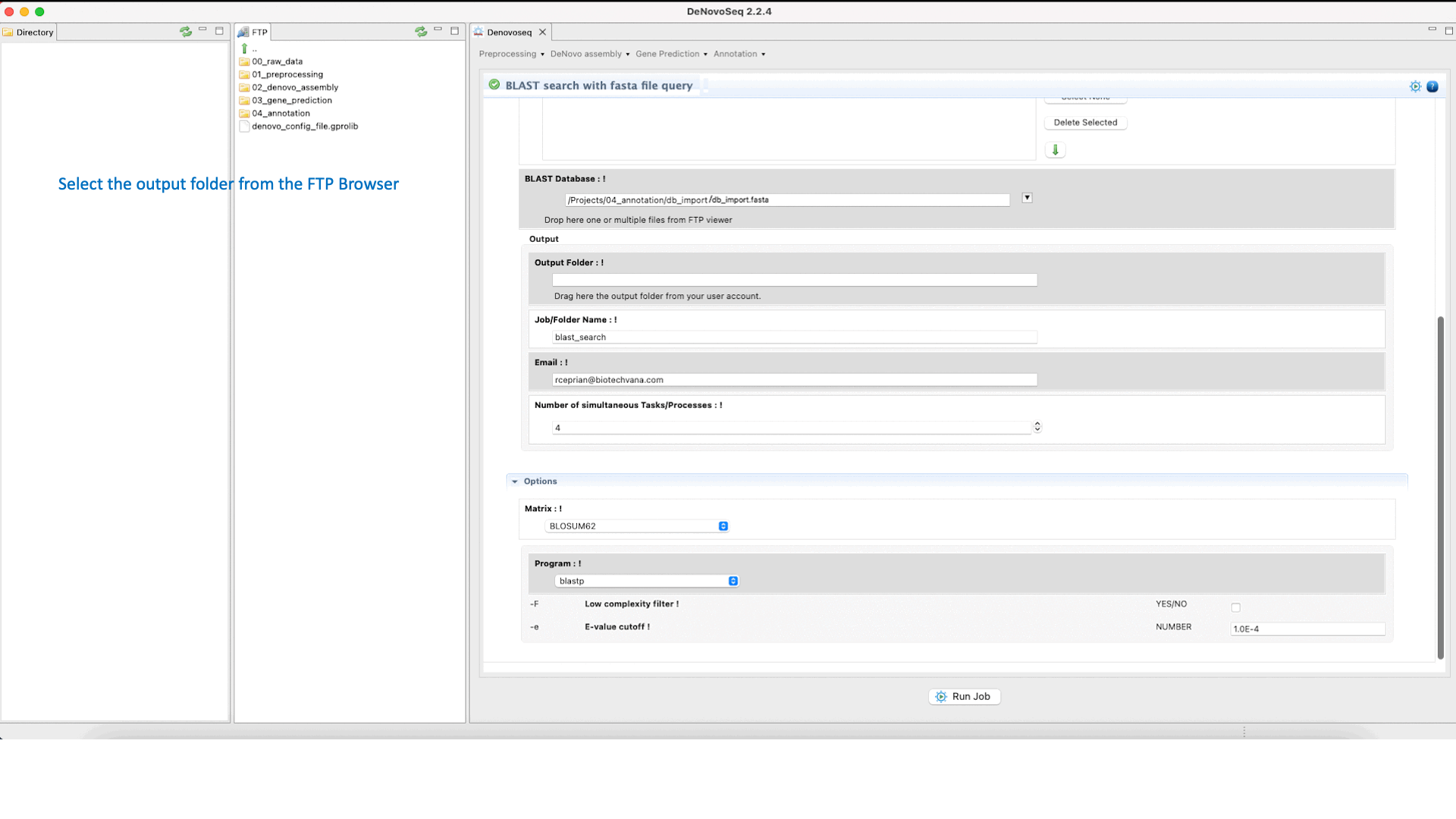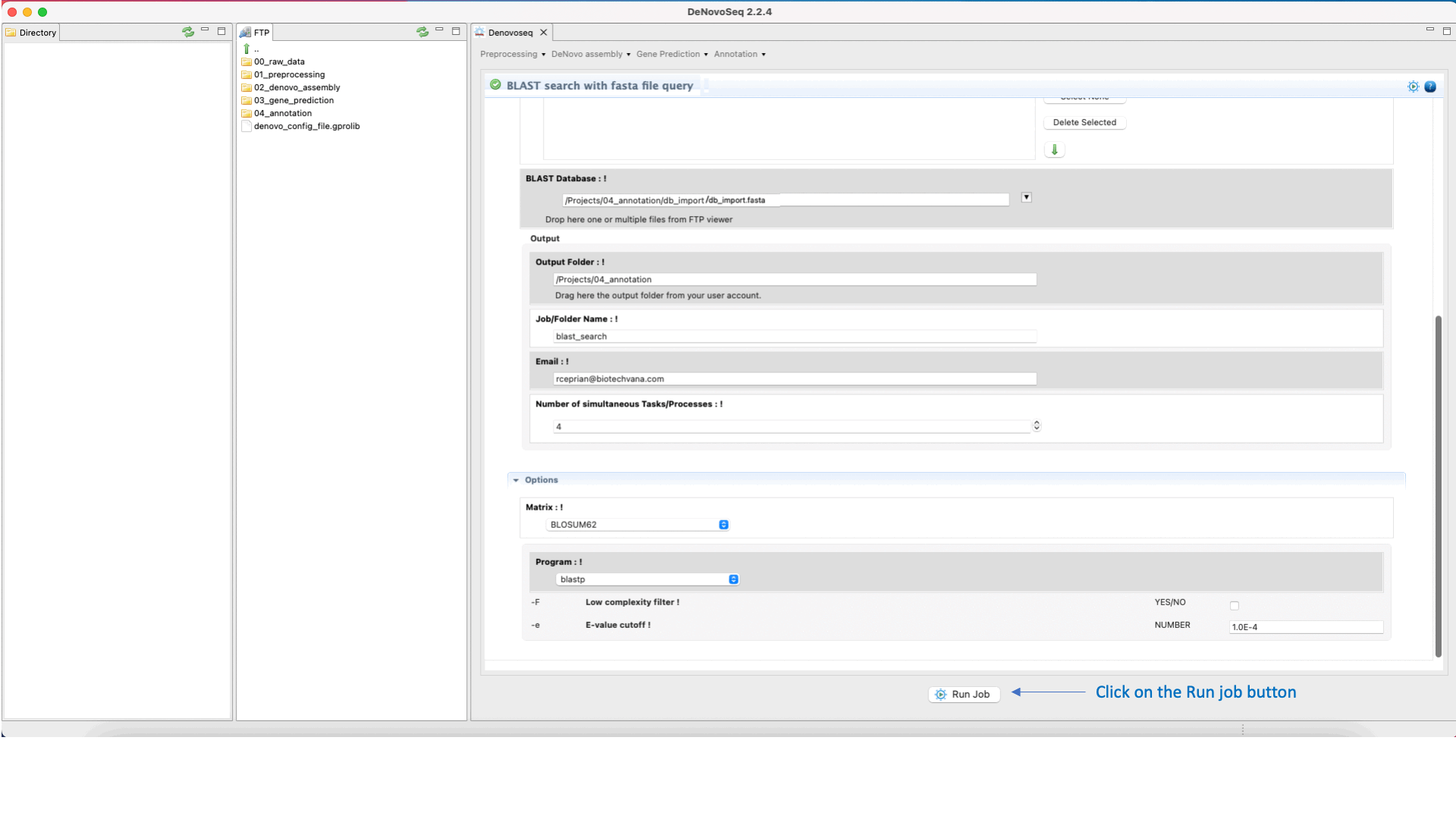
Figure 31: Using the GPRO interface to search blast-formatted databases with one or more queries using NCBI-BLAST package.
- Process BLAST output
The output of the BLAST search delivered by DeNovoSeq for multifasta files is a set of XML files (one per each sequence queried to the subject database) containing all matches obtained found in the database by each query. This thus means that if a multifasta file annotated via BLAST has 25000 sequences the BLAST search will report 25000 xml files. DeNovoSeq also provides an interface for an internal script of GPRO for automatic annotation. This script processes the XML outputs provided by BLAST and prints them into a human-readable annotation file in CSV format. The interface for this script provides filtering parameters to define an evalue cut-off, filter redundant matches, extract a given number of best hits per query and more.
To execute the process BLAST output script, go to [Annotation → NCBI-BLAST → Process BLAST outoput] and follows in Fig.32.
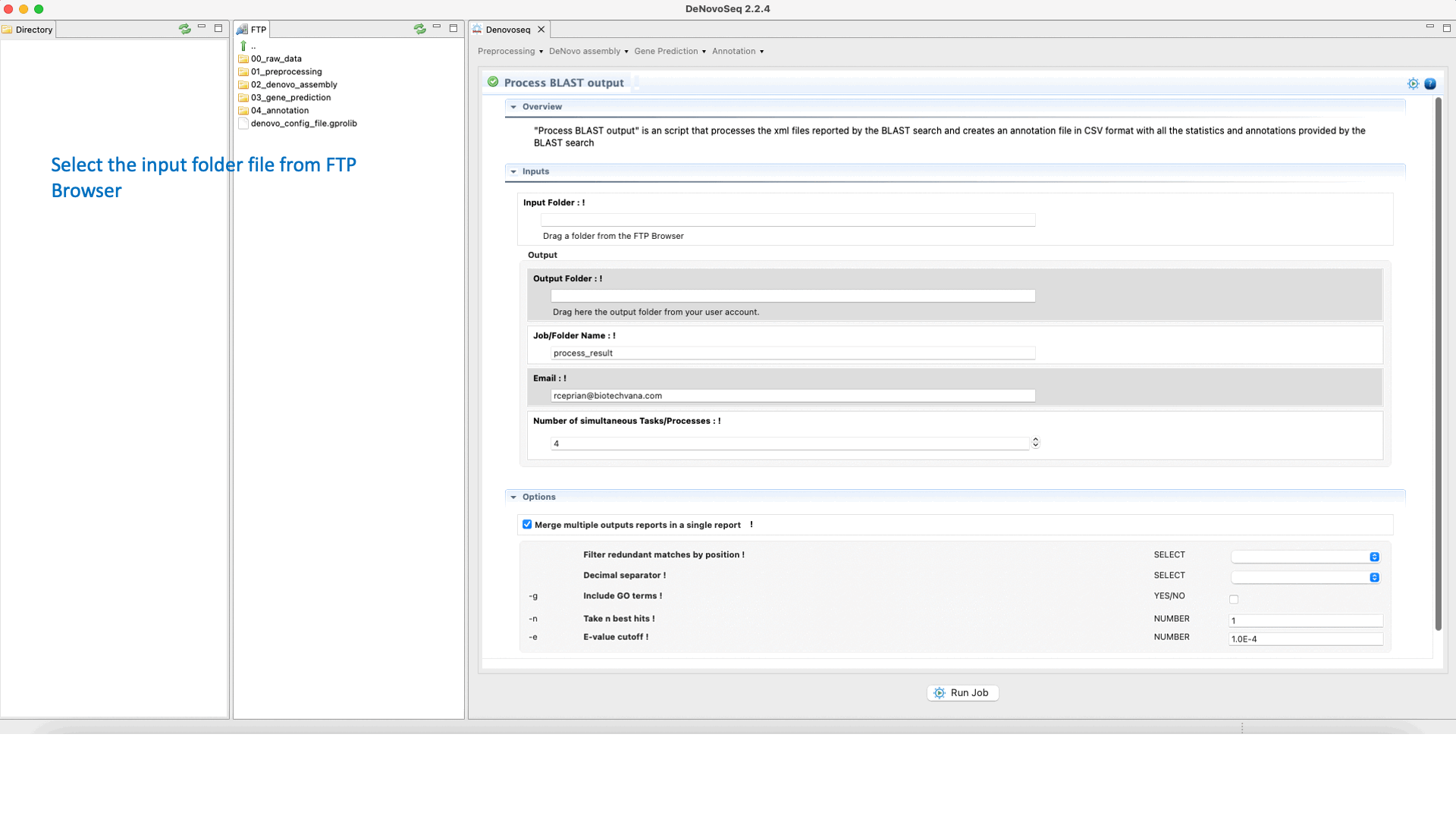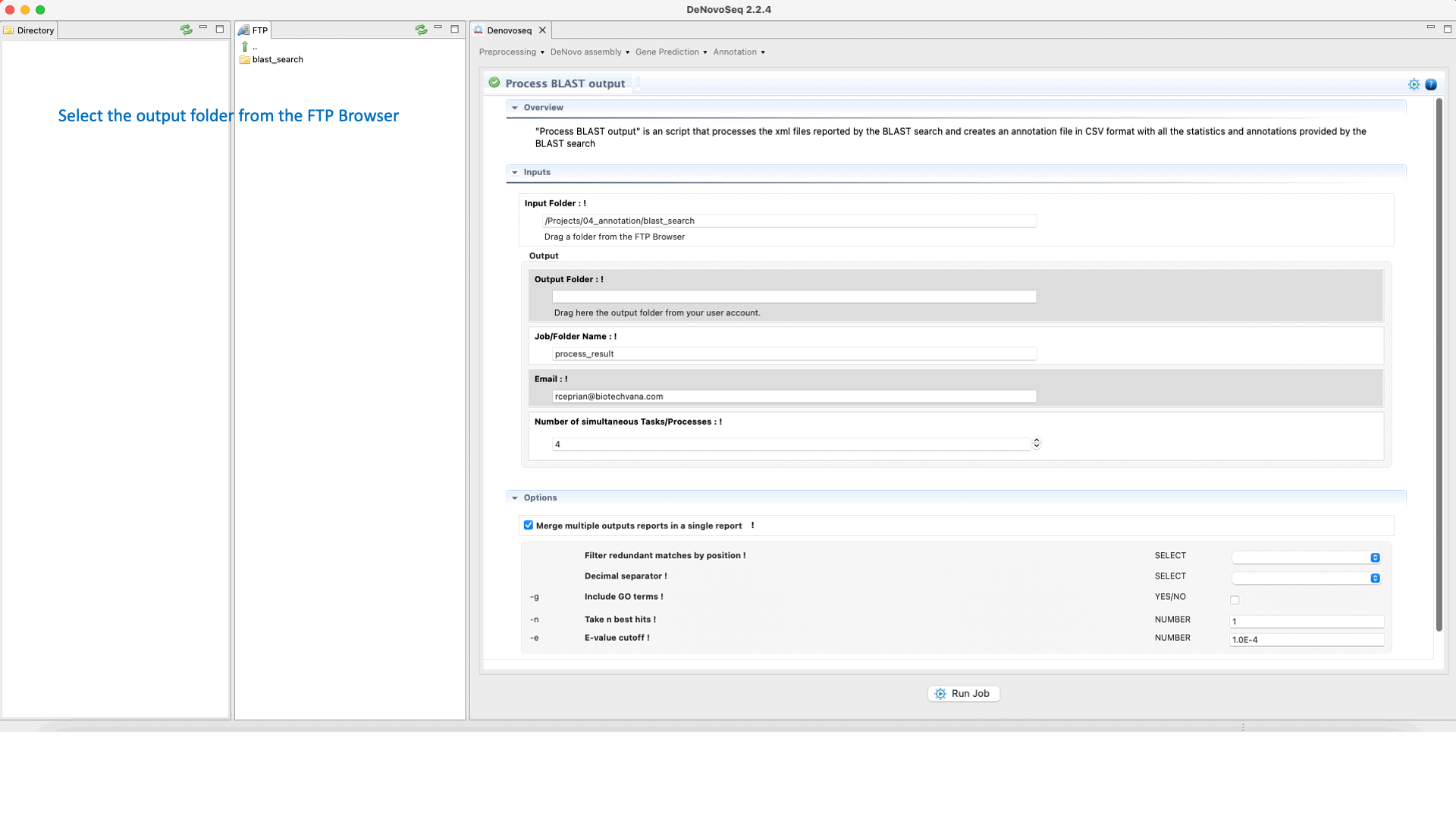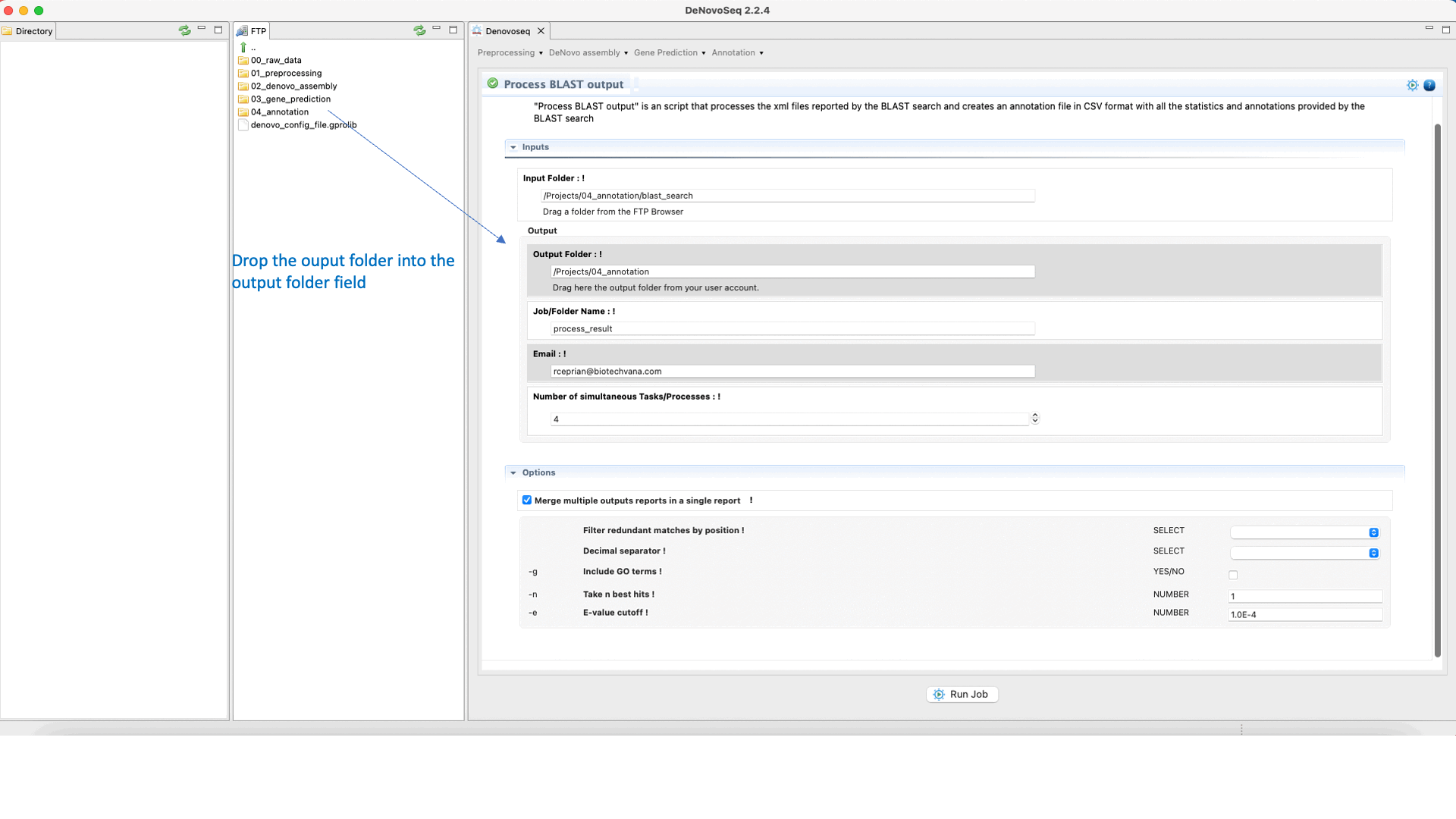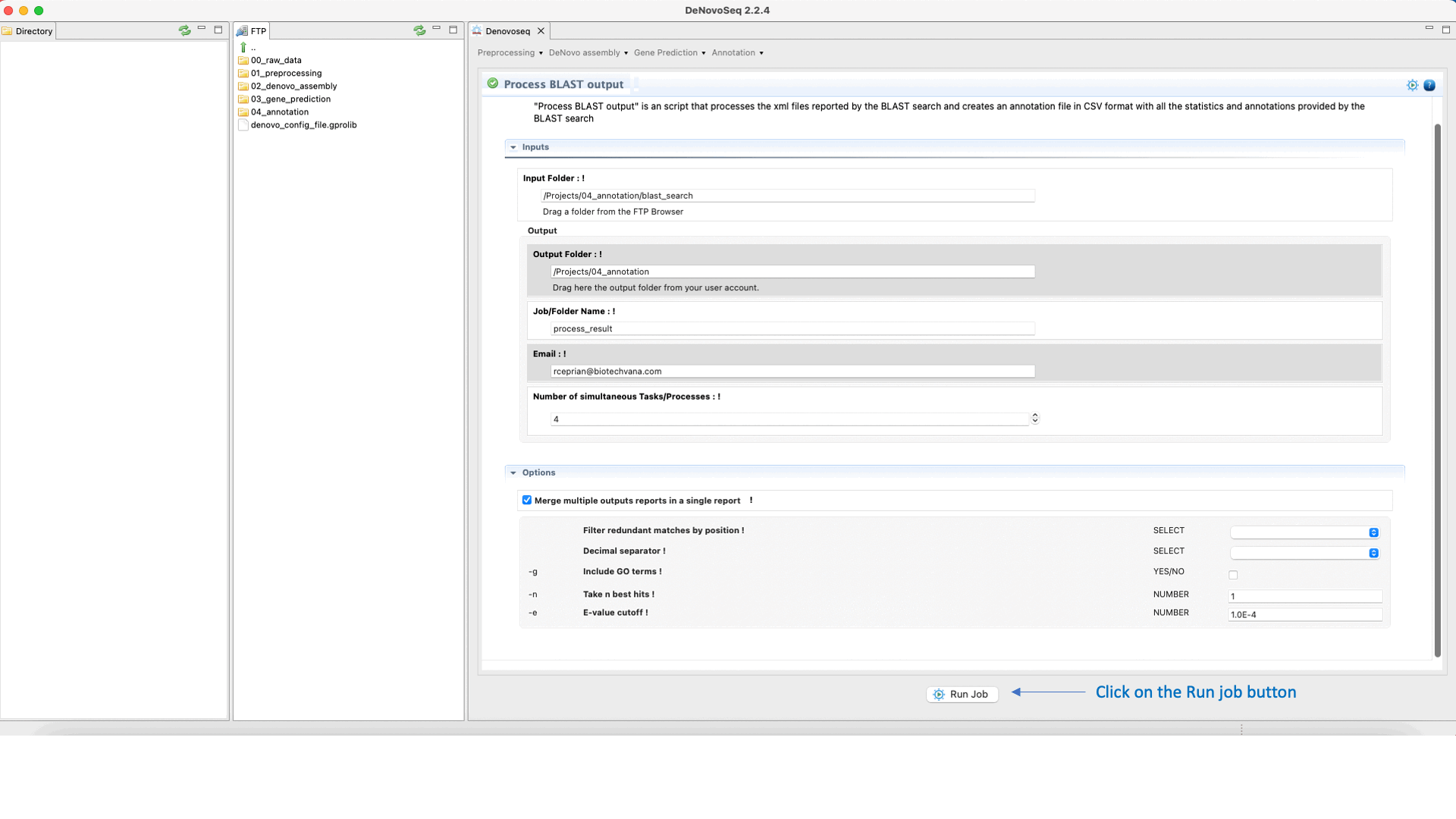
Figure 32: Using the GPRO interface for processing BLAST outputs and extracting annotation files from BLAST results.
- Retrieve sequences from BLAST outputs
DeNovoSeq also provides a parsing script for extracting sequences (that can be either from the query file or from the subject database) according to results provided by the BLAST search and create a new fasta file containing only with the sequences retrieved. The interface for this script also provides filtering parameters to extract full sequences or just the core of the query or subject sequence that aligns constituting the High-scoring Segment Pair (HSP). This last mode also permits to make extractions extending “n” nucleotides upstream and downstream of the HSP core.
To access the script for retrieving sequences from query or subject multifasta files according to the BLAST output, go to [Annotation → NCBI-BLAST → Retrieve sequences from BLAST outputs] and follows what stated in Fig.33 for this script.
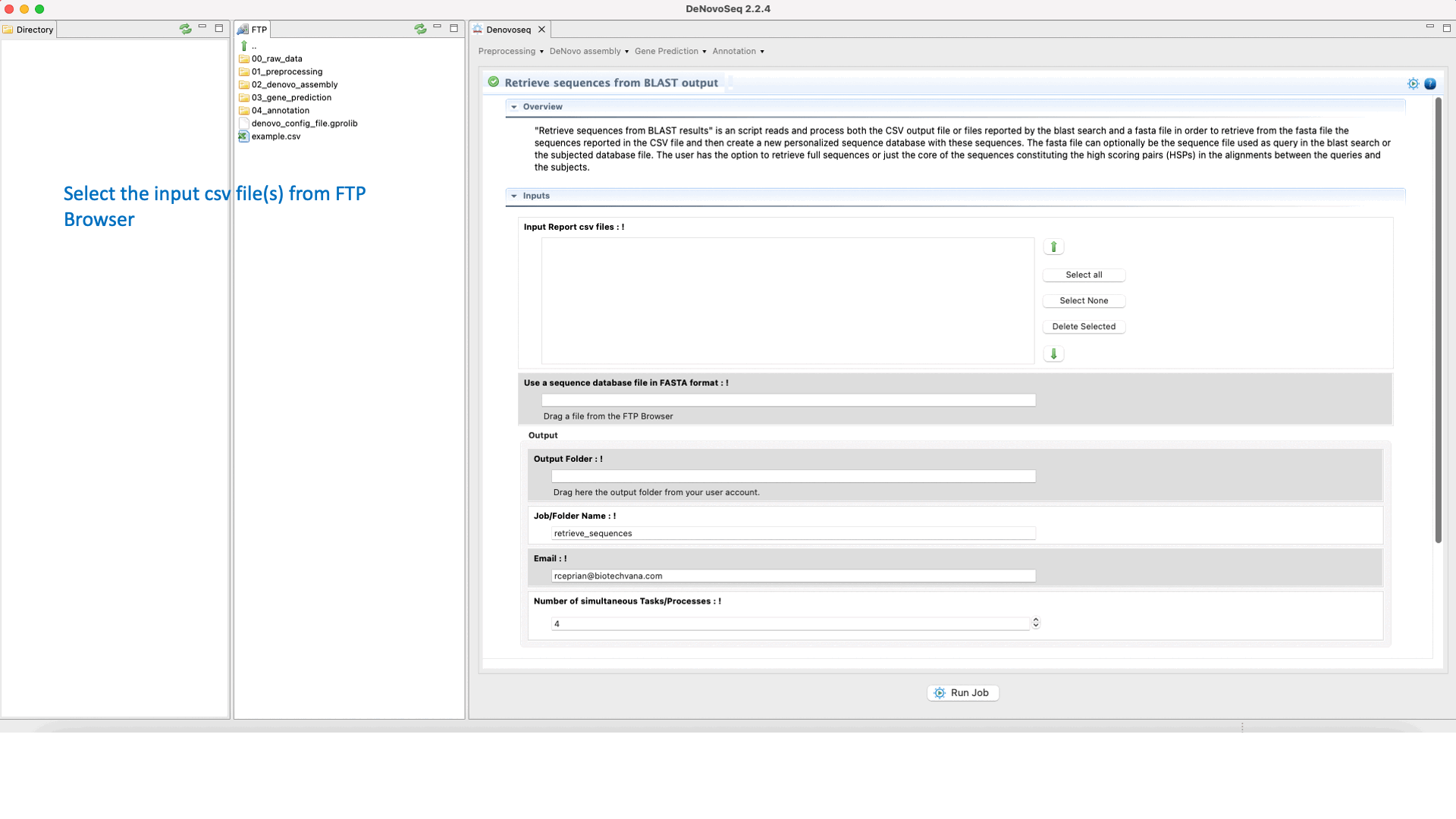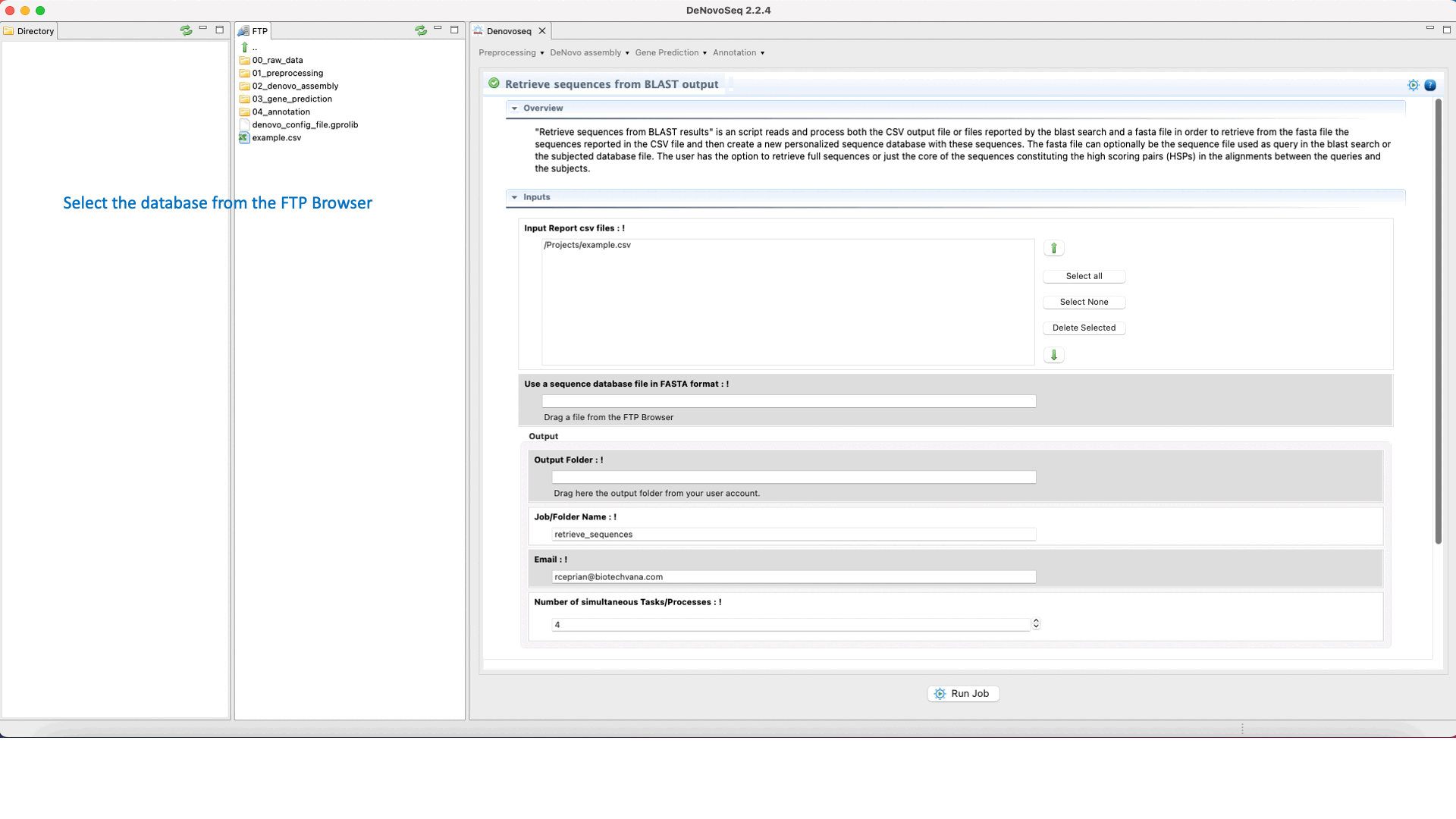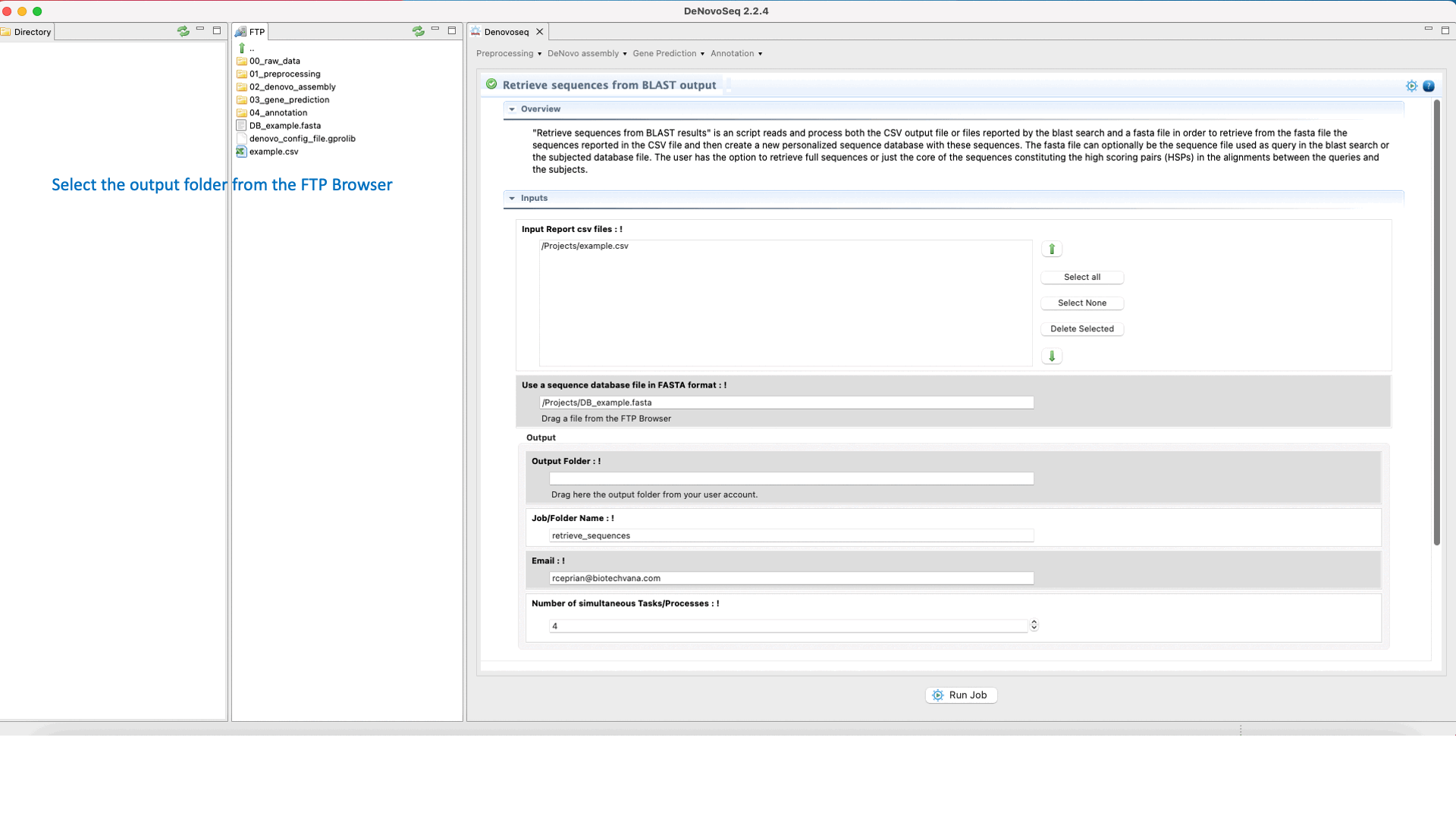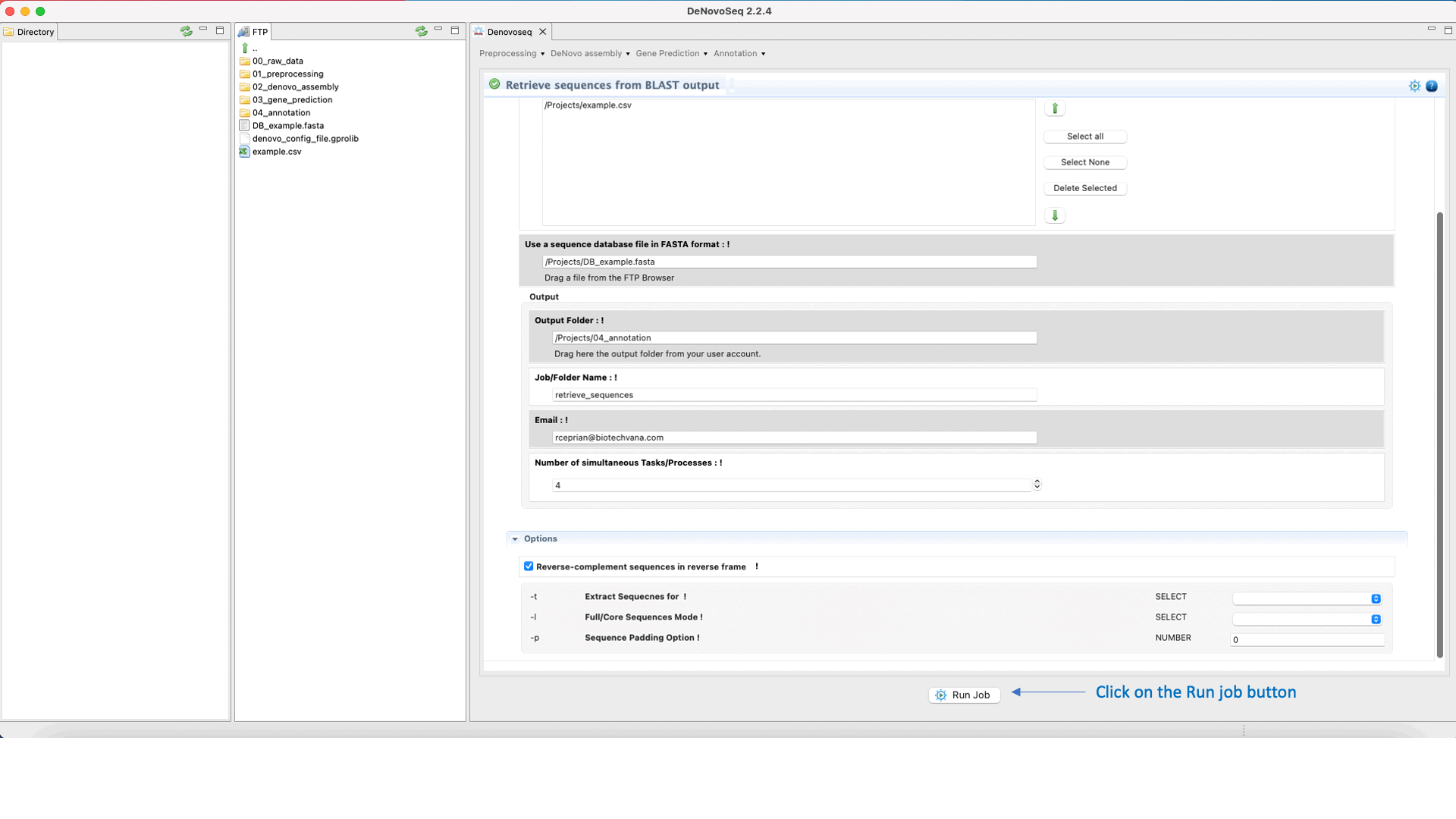
Figure 33: Using the GPRO interface for processing BLAST outputs and extracting sequences files from BLAST results.
HMMER3Mistry et al 2013is a software for searching sequence homologs that makes comparisons between protein or nucleotide sequence queries and a user-made database of Hidden Markov Model (HMM) profiles (or vice versa). HMM profiles are probabilistic models capturing position-specific information in a set of aligned sequences (i.e. a multiple alignment) about the evolutionary changes occurred per alignment position.
The step-by-step mode of DeNovoSeq presents different interfaces to manage HMMER for creating and editing HMM databases from multiple alignments, creating consensus sequences or for performing comparative analyses. See the manual of HMMER at "http://eddylab.org/software/hmmer/Userguide.pdf" for more information.
- Create HMMER databases
DeNovoseq provides an interface to call a small pipeline executing the HMMER commands hmmbuild, hmmcalibrate and hmmpress to respectively:
To execute the hmmbuild-hmmcalibrate-hmmpress pipeline go to [Annotation → HMMER → Create HMMER databases] and follows what stated in Fig.34 for creating HMMs.
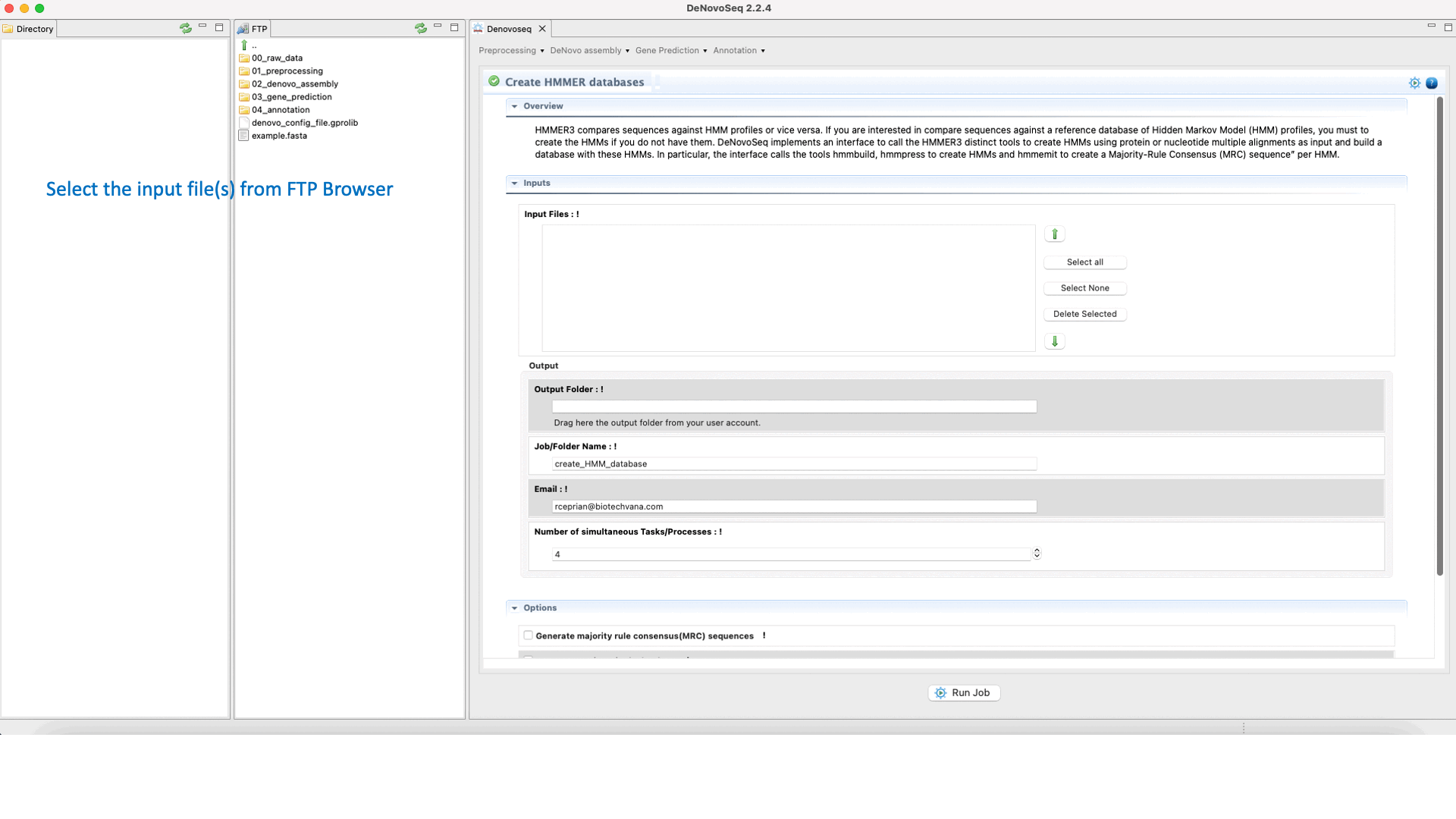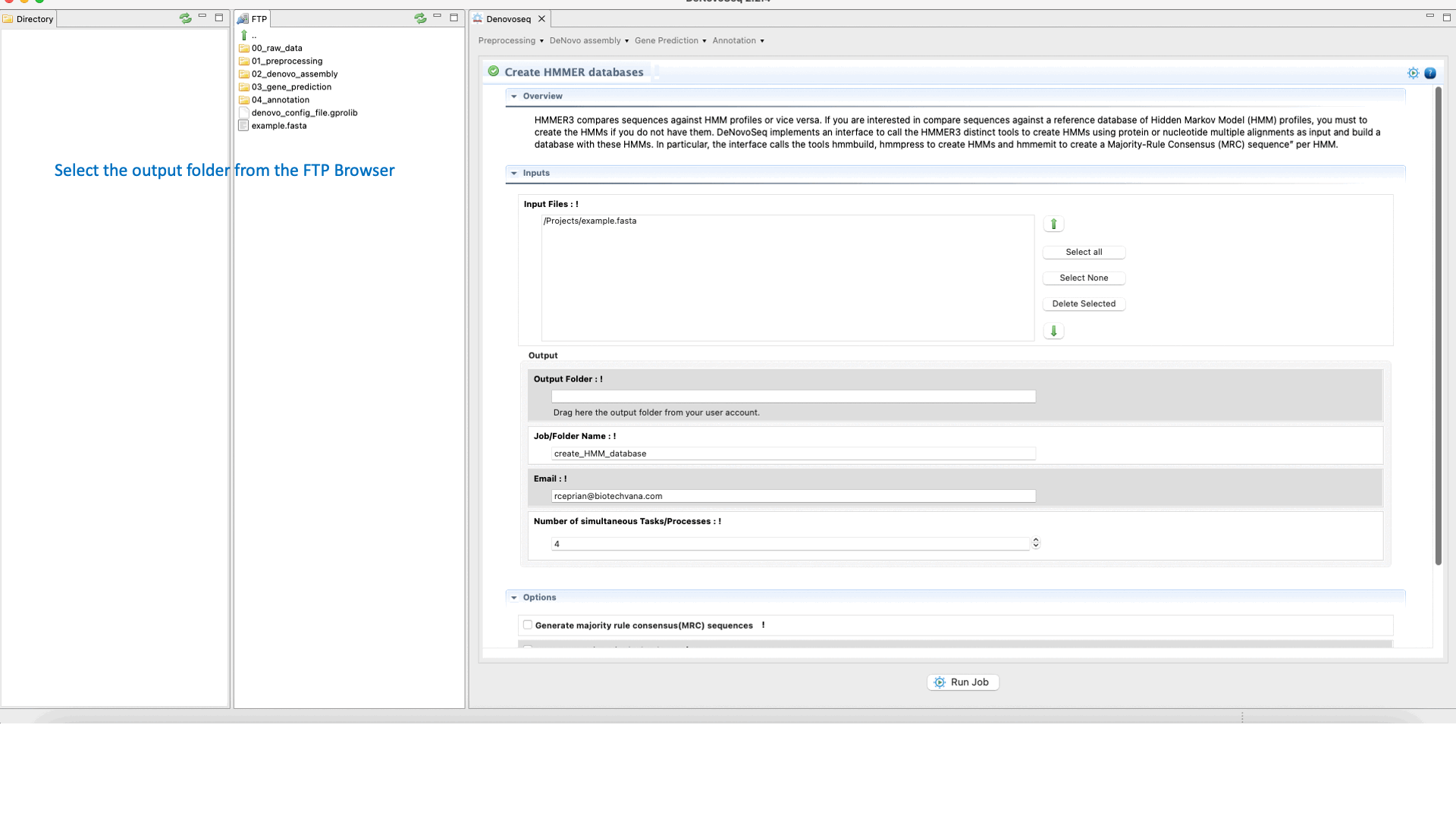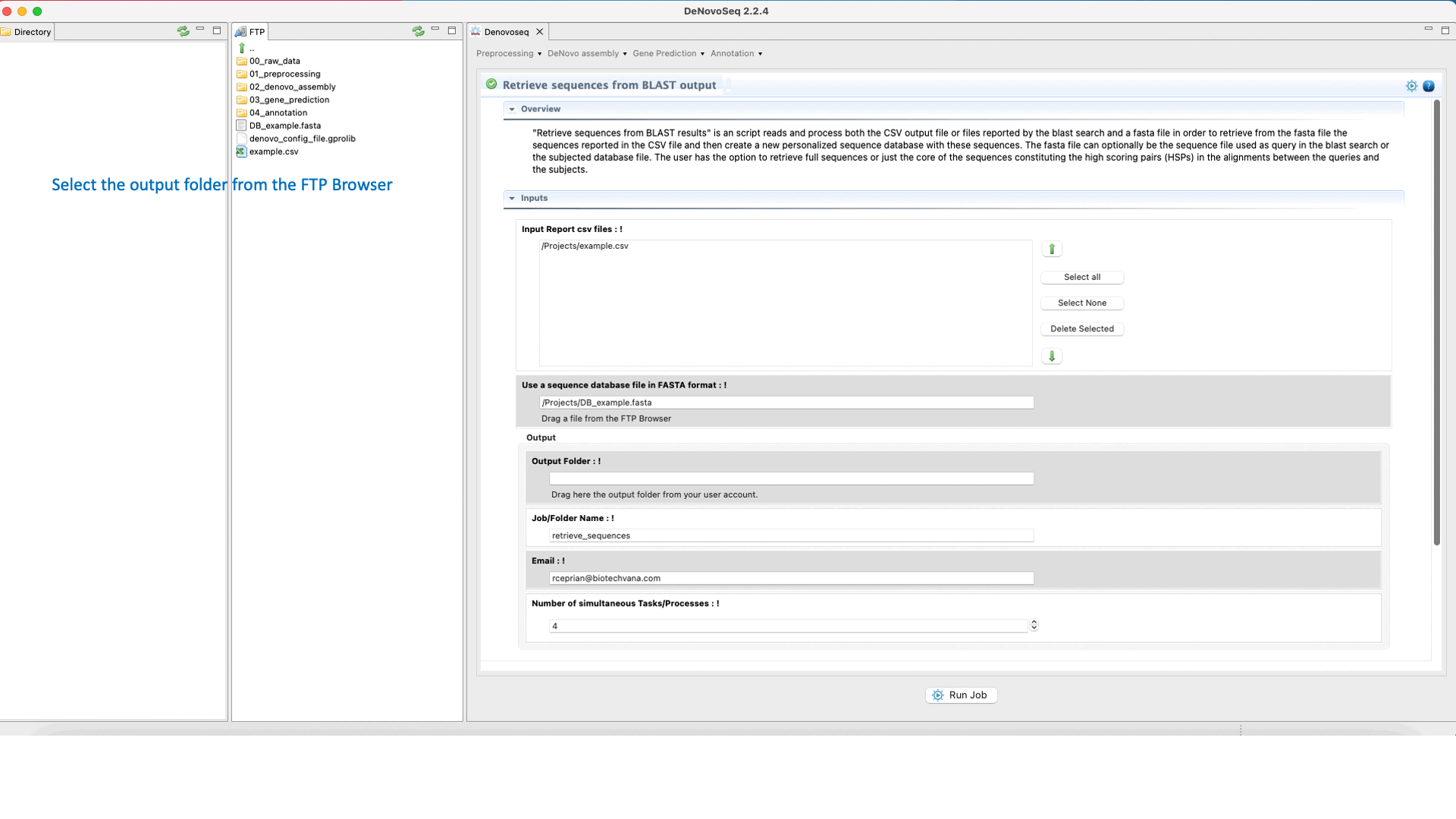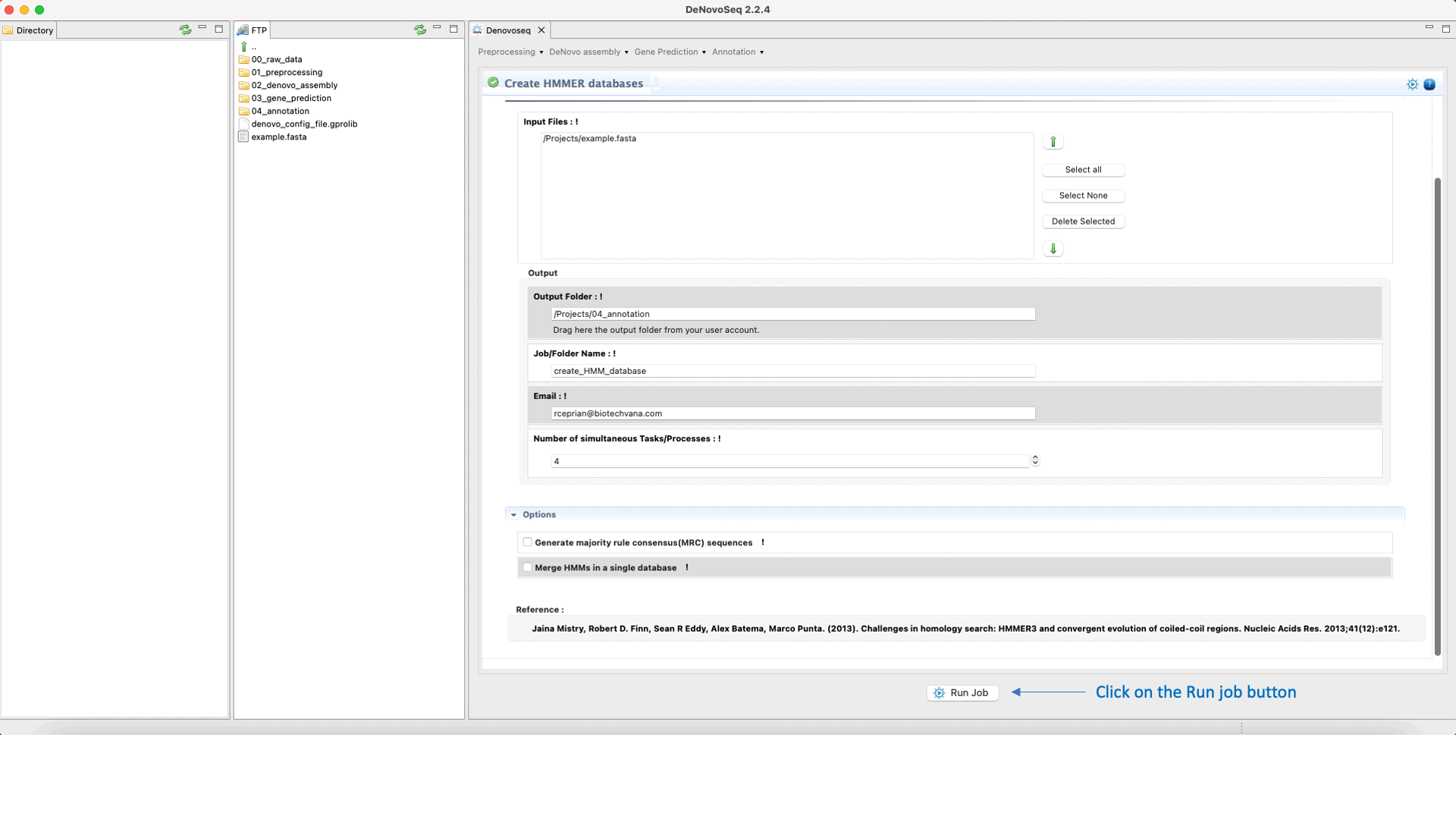
Figure 34: Using the GPRO interface for building HHMER databases.
- Edit HMMER databases
For editing HMM databases DeNovoseq provides an interface that calls the HMMER commands hmmalign and hmmemit to respectively:
To execute the hmmalign-hmmemit pipeline go to [Annotation → HMMER → Edit HMMER databases] and follows what stated in Fig.35 for editing and updating HMMs.
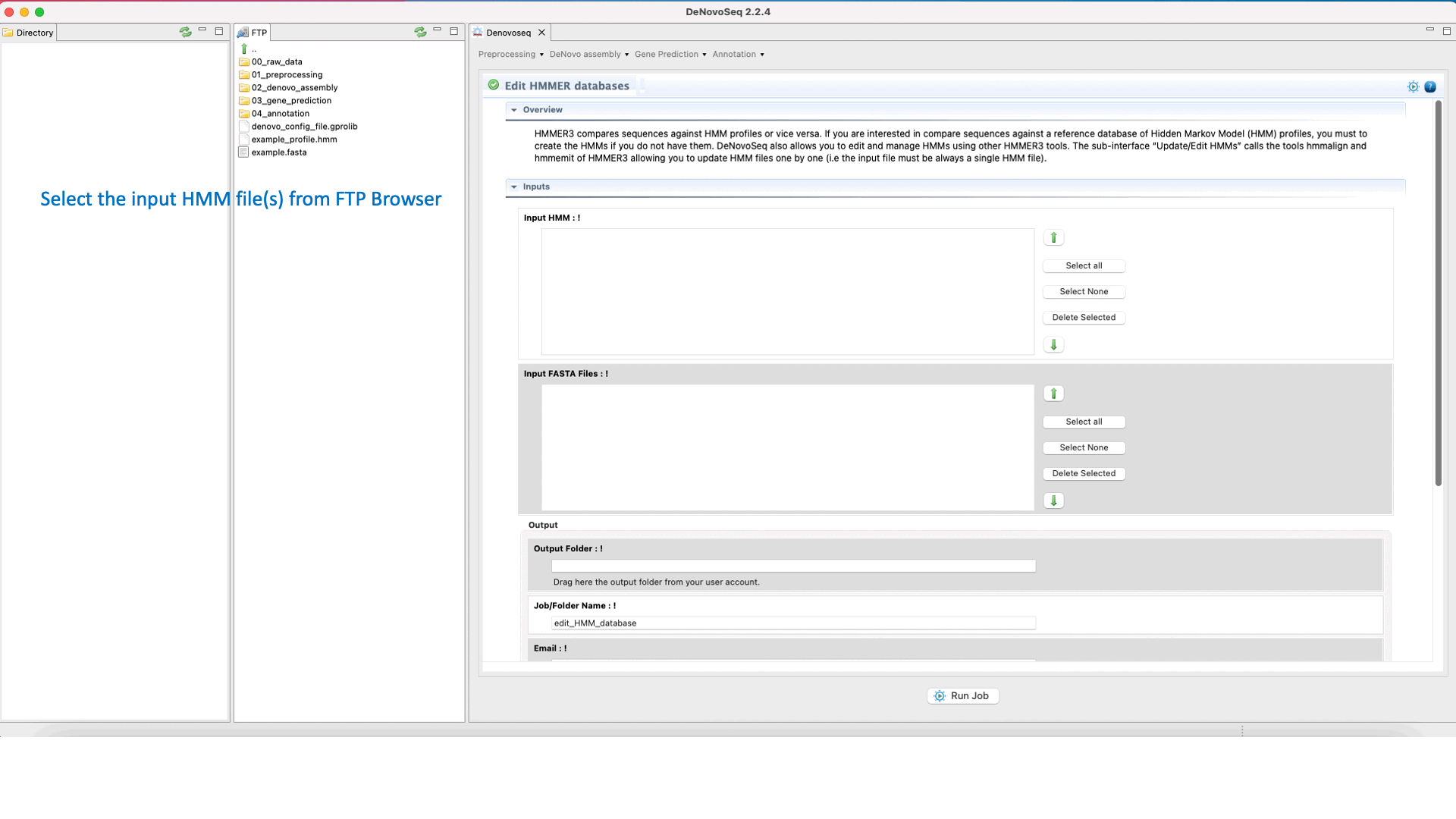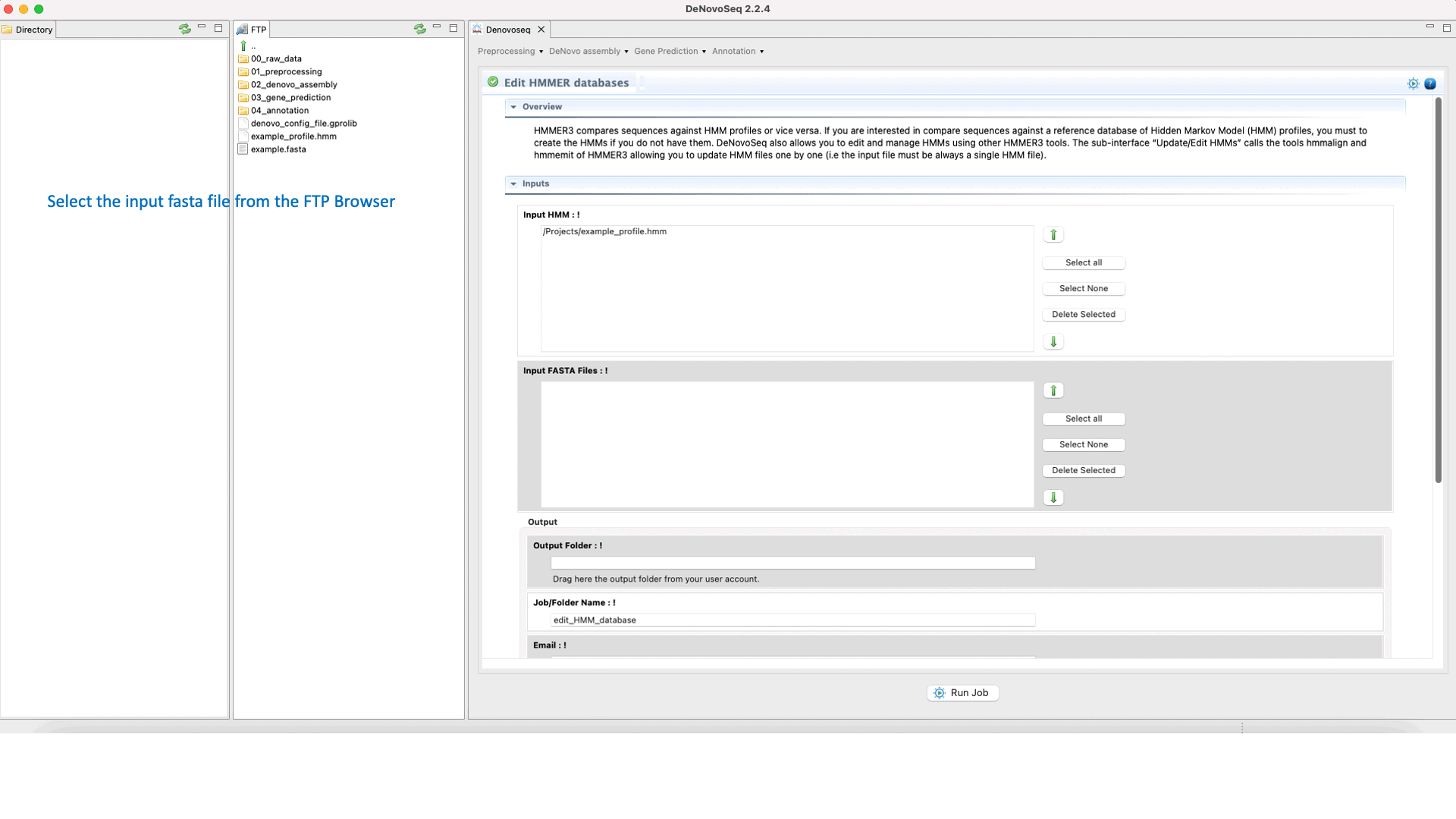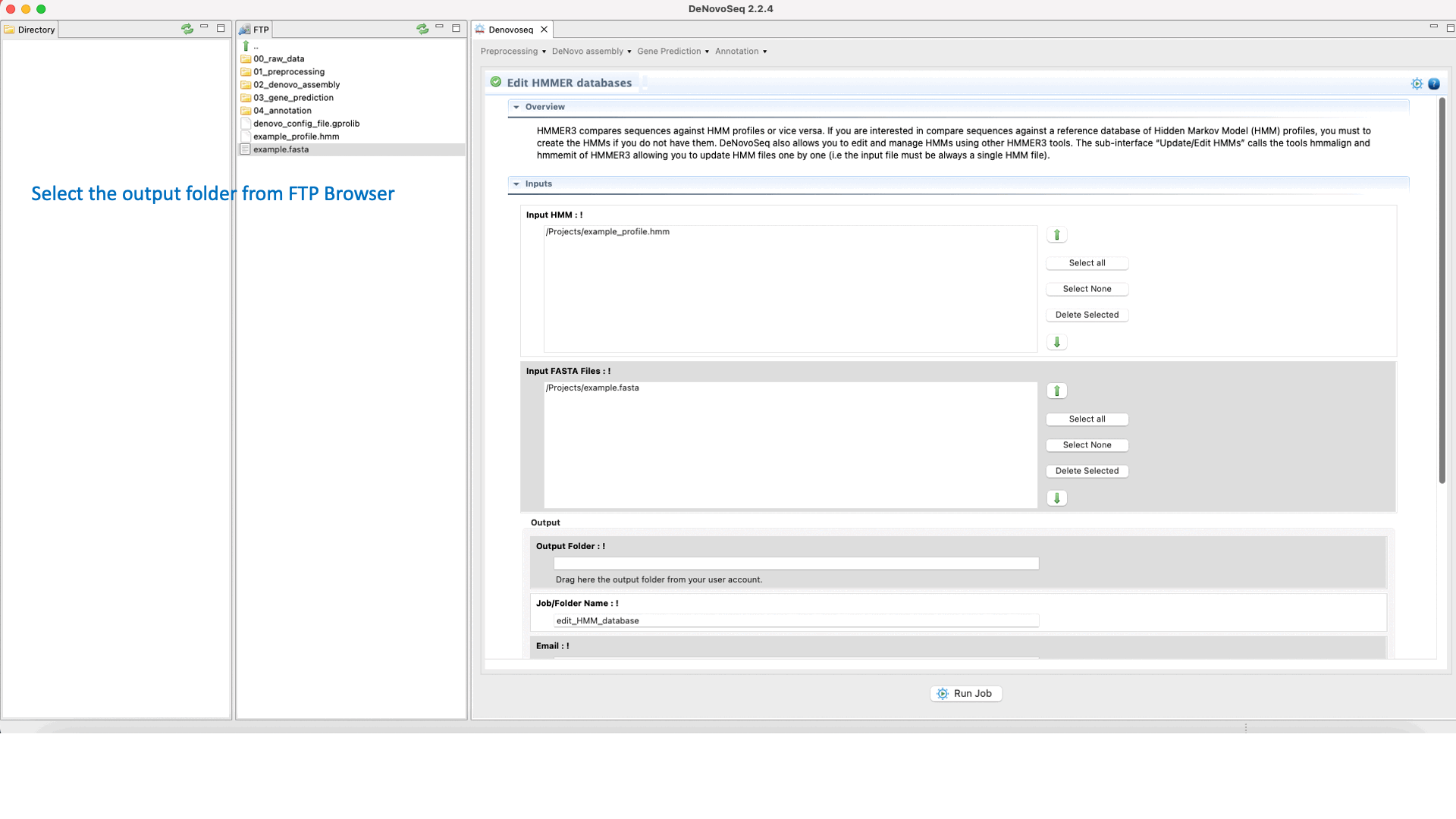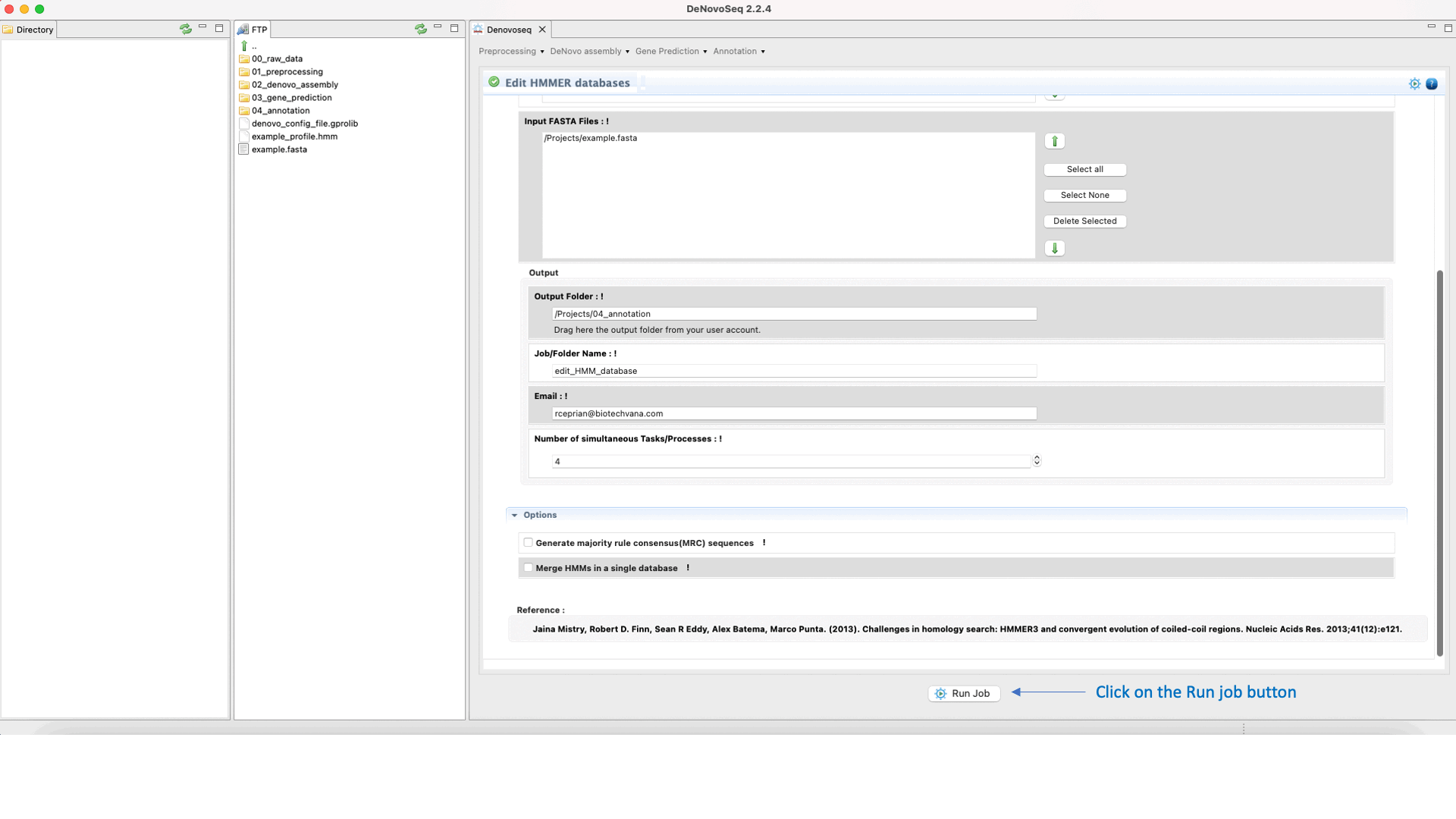
Figure 35: Using the GPRO interface for editing HHMER databases.
- HMMER search fasta file query
DeNovo Seq provides a frame for a web-based interface to run fast searches against the HMM databases using one sequence or multiples queries and obtaining the typical alignment output provided by HMMER. Three kinds of HMM searches are allowed:
For annotation of multifasta files, DeNovoSeq provides a specific interface allowing the users to run multiple searches with HMMER3 executed in background process. For large query files the interface also permits the user to divide the input file in multiple subqueries to accelerate the process.
To execute a HMMER search for multifasta file queries, go to [Annotation → HMMER → HMMER search with fasta file query] and follows what stated for multifasta files searches in Fig.36.
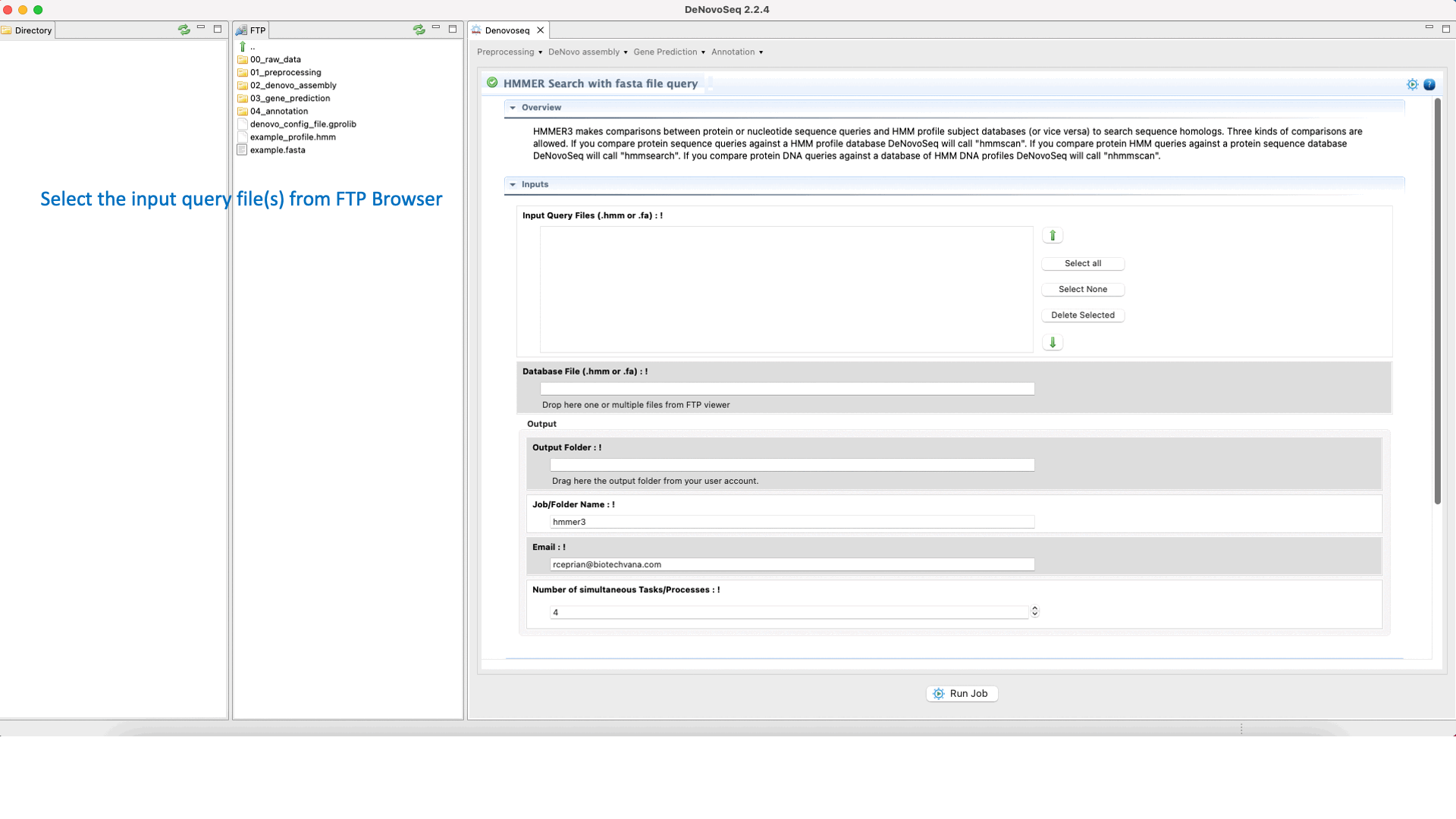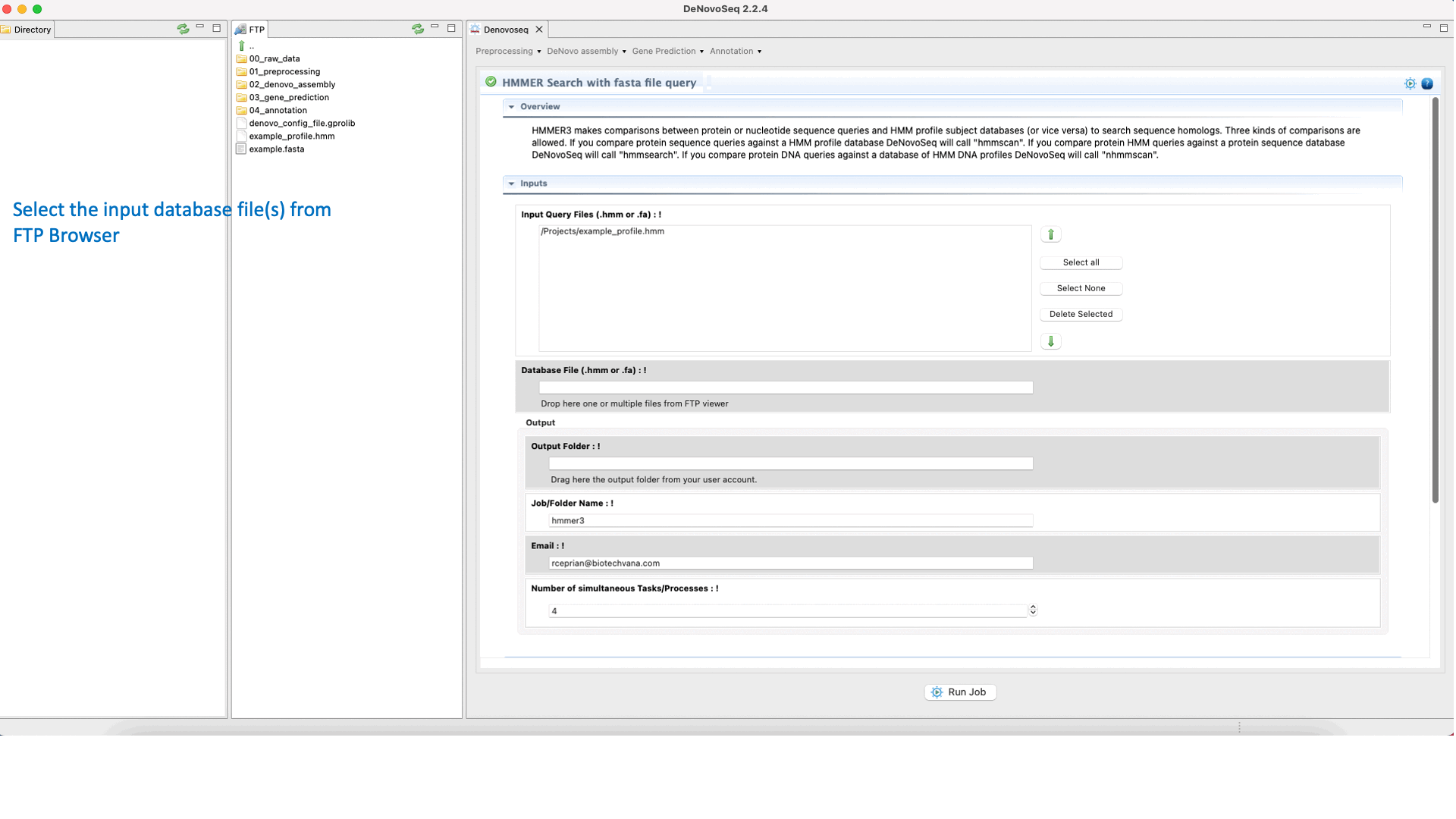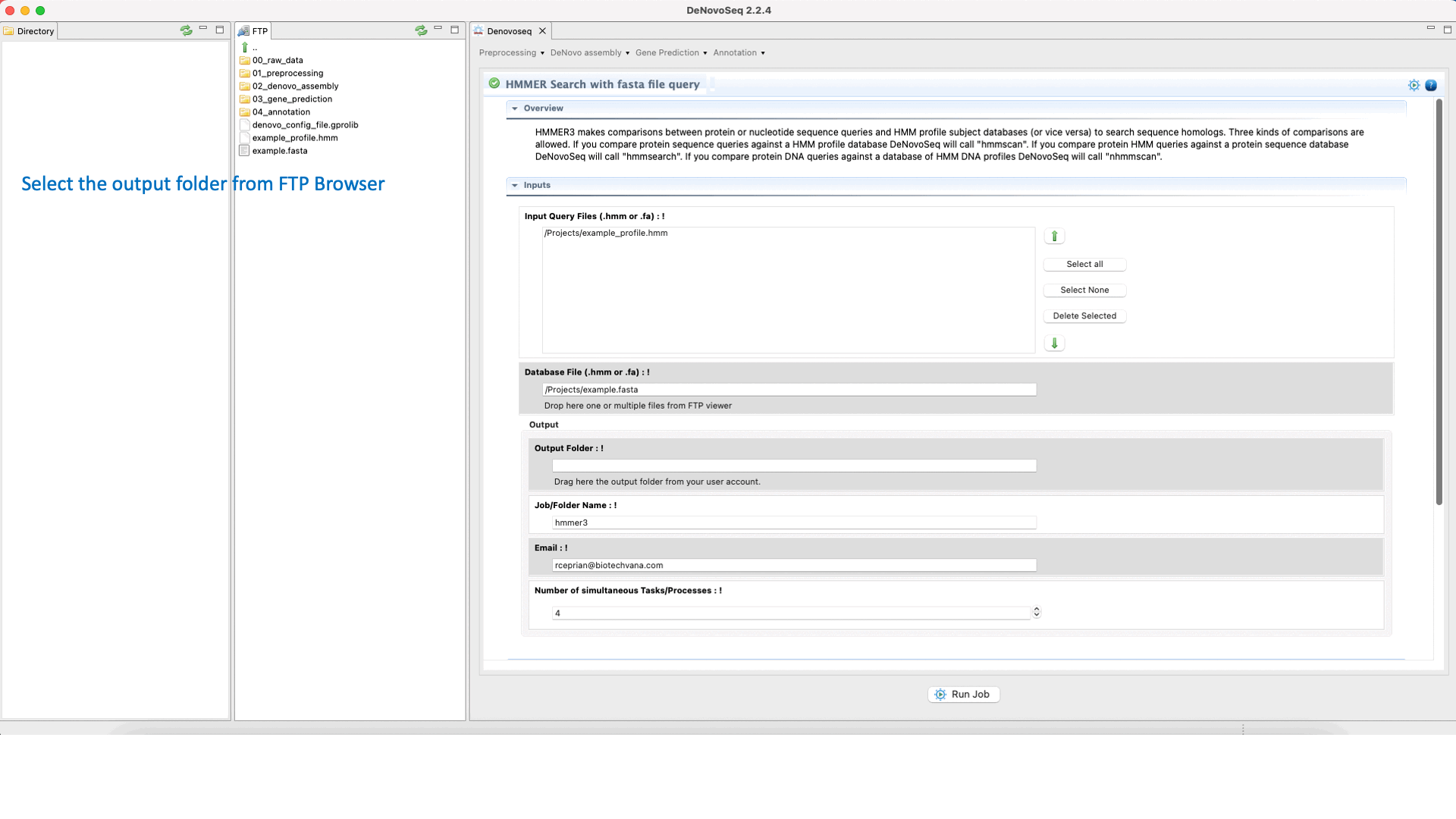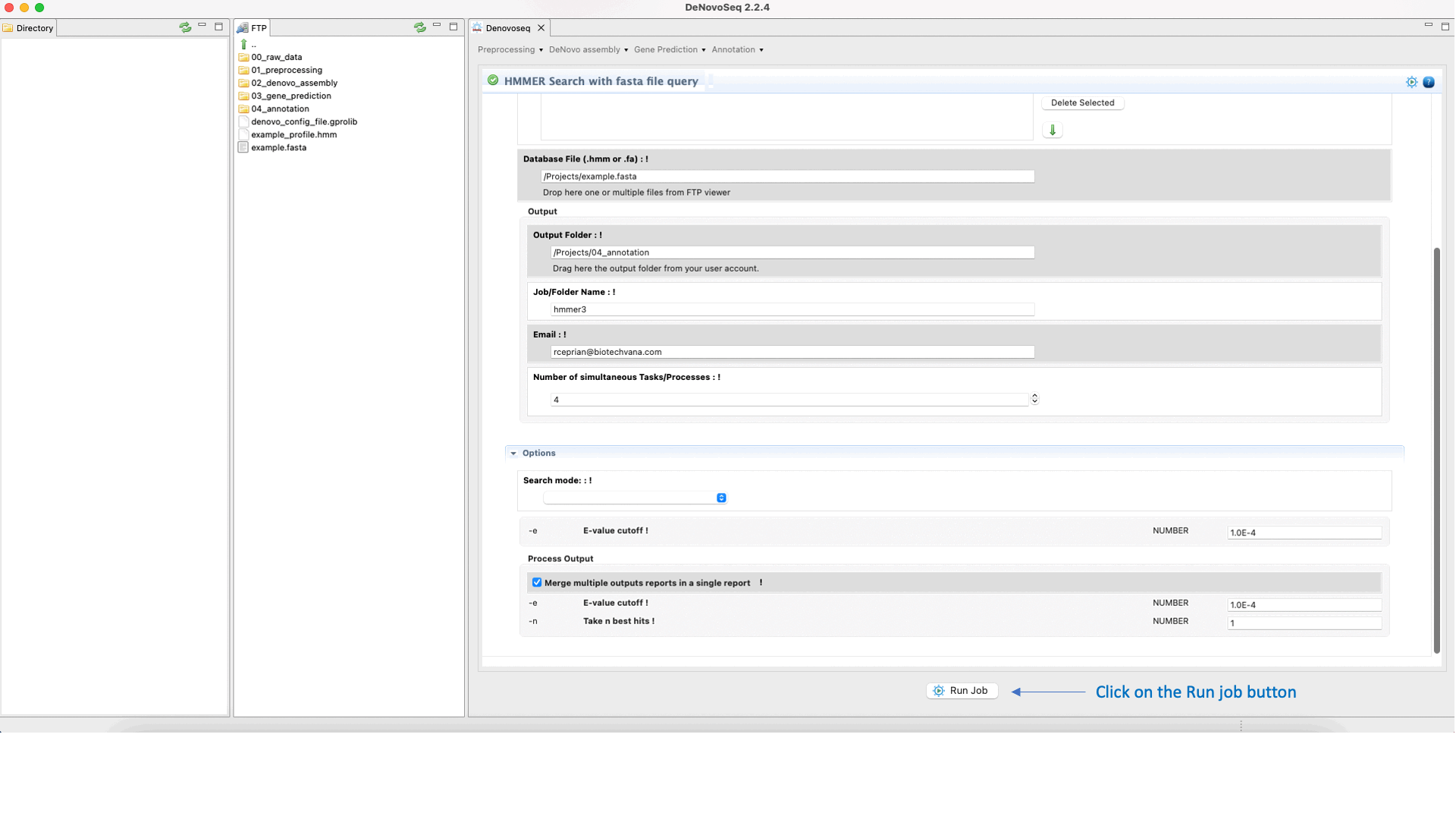
Figure 36: Using the GPRO interface for searching against the HMM databases using one sequence or multiples queries and obtaining the typical alignment output provided by HMMER.







