VariantSeq is a Client-Desktop Application of the GPRO suite that uses best practices for analysis of Single Nucleotide Polymorphisms (SNP) and indels from DNA and VariantSeq data using the State-of-the -Art protocols (Fig.1). Tools for analysis of CNVs and/or structural variants are not yet implemented in the protocol. The application is an interface-based solution coupled with an infrastructure of server side dependencies (pipelines, databases and tools) that we distribute in a docker container that can be installed on a remote server or on a PC with sufficient RAM. The application also includes a File Transfer Protocol system (FTP) to facilitate the upload and download of files from the user’s computer to or from the server; a progress tracker (job tracking system), and two different execution modes (a “step-by-step” mode, and a “pipeline-like”mode).
The Step-by-Step mode is a procedure similar to those implemented in Galaxy (Afgan et al 2018),and others GUI-based solutions for NGS data analysis. This mode enables the user to run the analyses as a workflow of steps where each step can be completed independently from all other steps and where the options and the parameters of each analysis are declared prior to run the analysis. The Step-by-Step menu organizes the different steps of the variant analysis workflow (i.e. quality analysis, preprocessing, postprocessing, mapping, variant calling, filtering, annotation) into an intuitive menu providing a tab per step and a scroll down per tab summarizing the command line interface (CLI) software available for each step. In that way, the Step-by-Step mode permits the user to manage different protocols for variant analysis depending on the topic to address (populational studies, cancer, mendelian or hereditary diseases etc).
In contrast, the pipeline mode is a pipeline configuration system allowing the user to execute all the steps of a given protocol automatically one after the other. To this end the user just need to select a specific pipeline from a list, declare the experiment design as well as the input and output data, then configure the option and parameters and finally run the pipeline where the distinct analyses will be executed sequentially one after another.
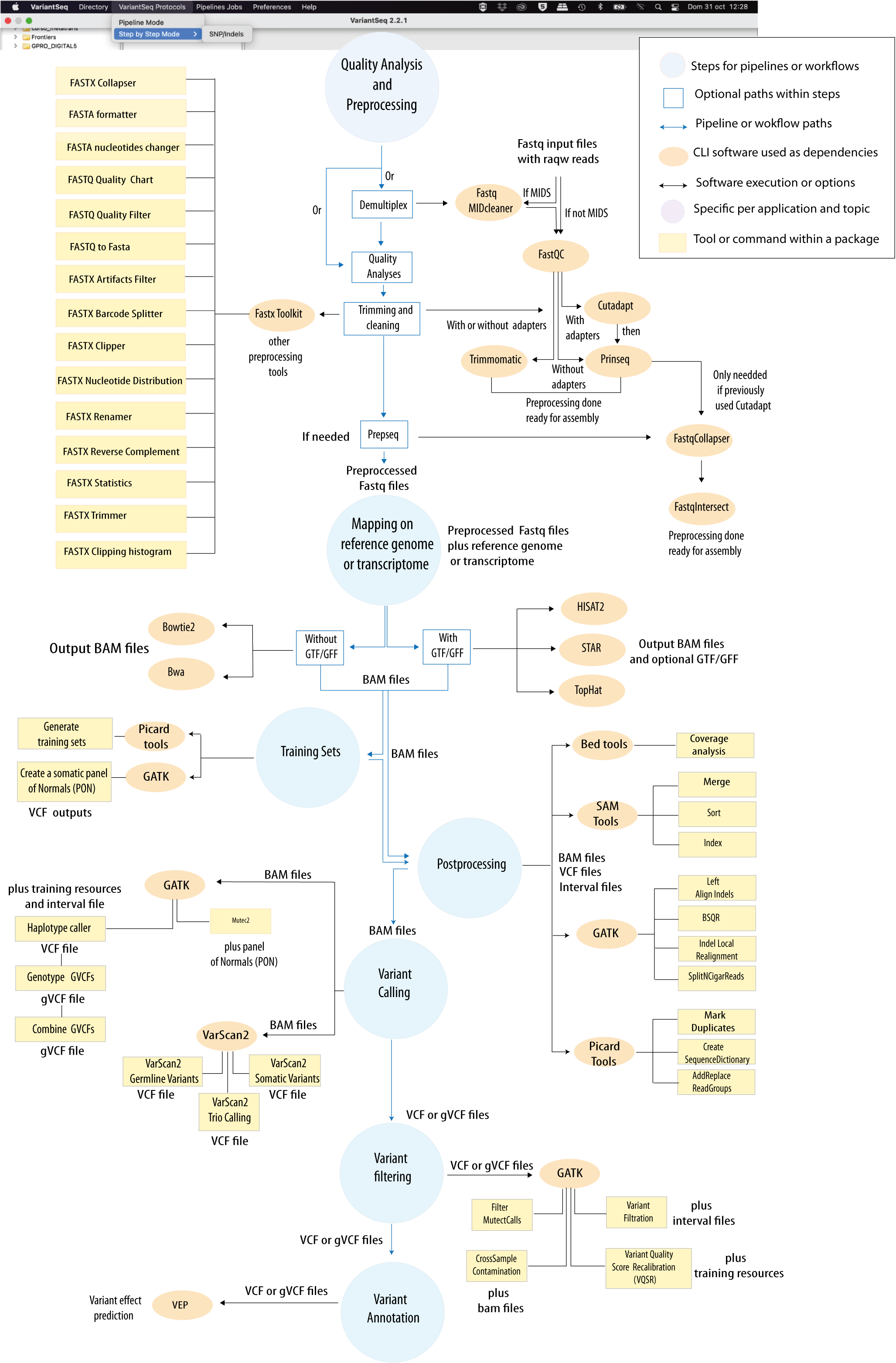
Figure 1: Bioinformatic protocol implemented in VariantSeq for analysis of Single Point Mutations and Indels according to the most common Variant-seq practices. The tool provides two execution modes (Step-By-Step and Pipeline-like) for execution of this protocol. An interactive scheme of this protocol is available at GENIE our virtual assitant.
java -version |
$ java -version
|
VariantSeq-win32.win32.x86_64.zip file and unzip it.
Then browse to the executable file "VariantSeq.exe" and execute/run it.
VariantSeq-macosx.cocoa.x86_64.zip file and unzip it
Then browse to the executable binary file "VariantSeq.app" and execute/run it.
VariantSeq-linux.gtk.x86_64.zip file and unzip itVariantSeq is a Client Side + Server Side solution thus meaning that the application is coupled via API with a bioinformatic infrastructure called GPRO Server Side that contains all the dependencies needed by VariantSeq to execute the workflows and pipelines. These dependencies are scripts, databases and the following third party CLI software:
The GPRO Server Side can be installed in the PC of the user or in remote servers as a Cloud Computing resource. However, its installation is a complex task due to the lot of dependencies and requirements (besides of the CLI software) for installing and running this infrastructure. For this reason, we distribute the GPRO Server Side in a Docker container that can be easily installed for the user in a couple of steps. Indications for installation of the GPRO Server Side Docker are available here.
Once the GPRO server side docker has been installed you need to link VarianSeq to it. To do this, go to [Preferences → Pipeline connection settings] in the top menu and type the following into the configuration Dialog (Fig.2):
As also shown in Figure 2 you can also check the option “Run GPRO server locally using Docker” to let you to automatically start the GPRO container each time you run VariantSeq (Also note that if you have this option checked you do not need to type the port). You can test if the app is connected to the Server Side clicking on the tab “Test connection settings”. Alternatively, if you install the Server Side manually (without the Docker) just add the IP of the remote server where the Server Side is hosted, add the port information (by default 22) and keep the Option “Run GPRO server locally using Docker” unchecked
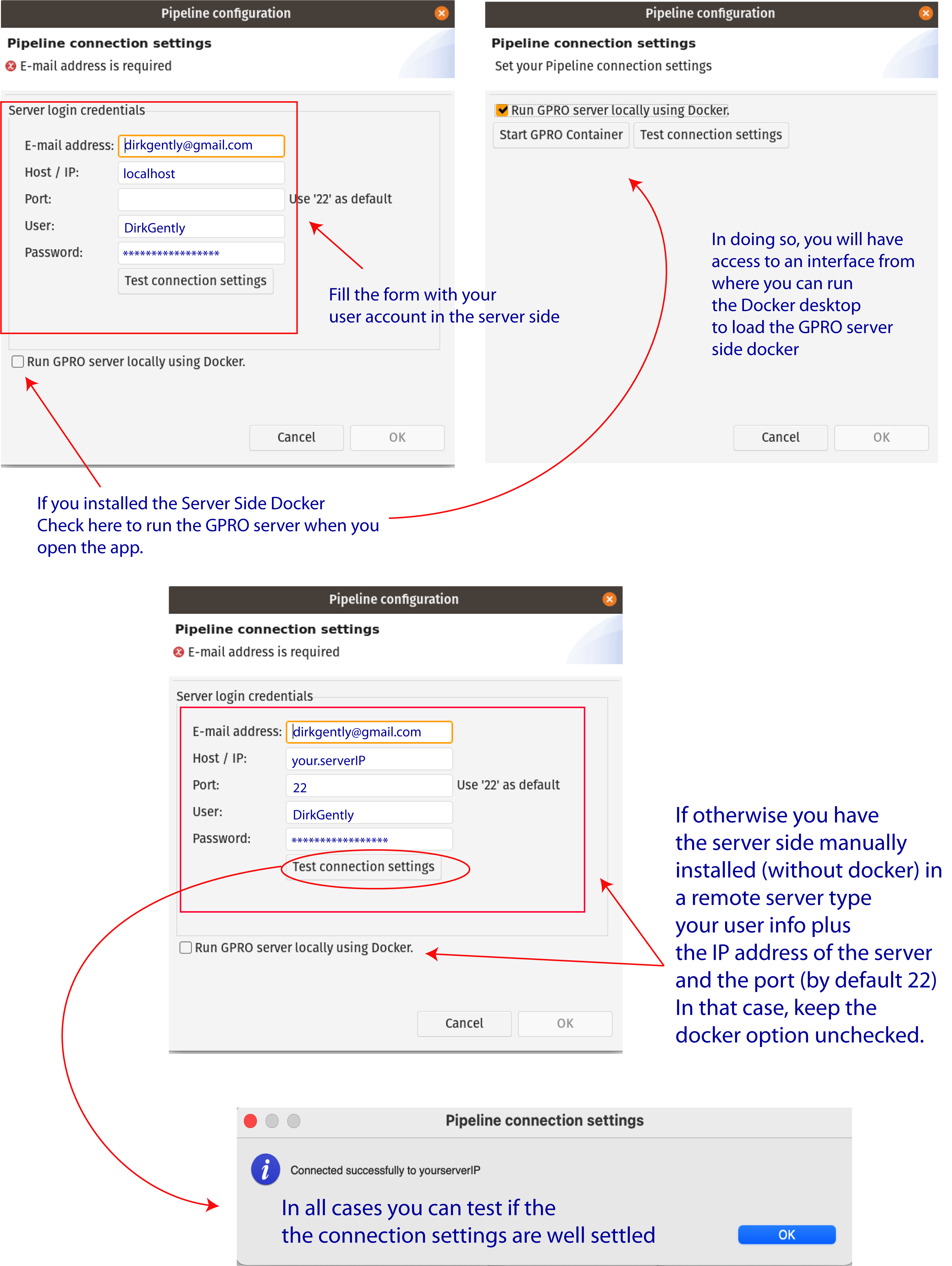
Figure 2: Server connection dialog.
[VariantSeq.app → Show package contents → Contents → MacOS → VariantSeq.ini]
Xms1024m (Minimum allocated memory)
|
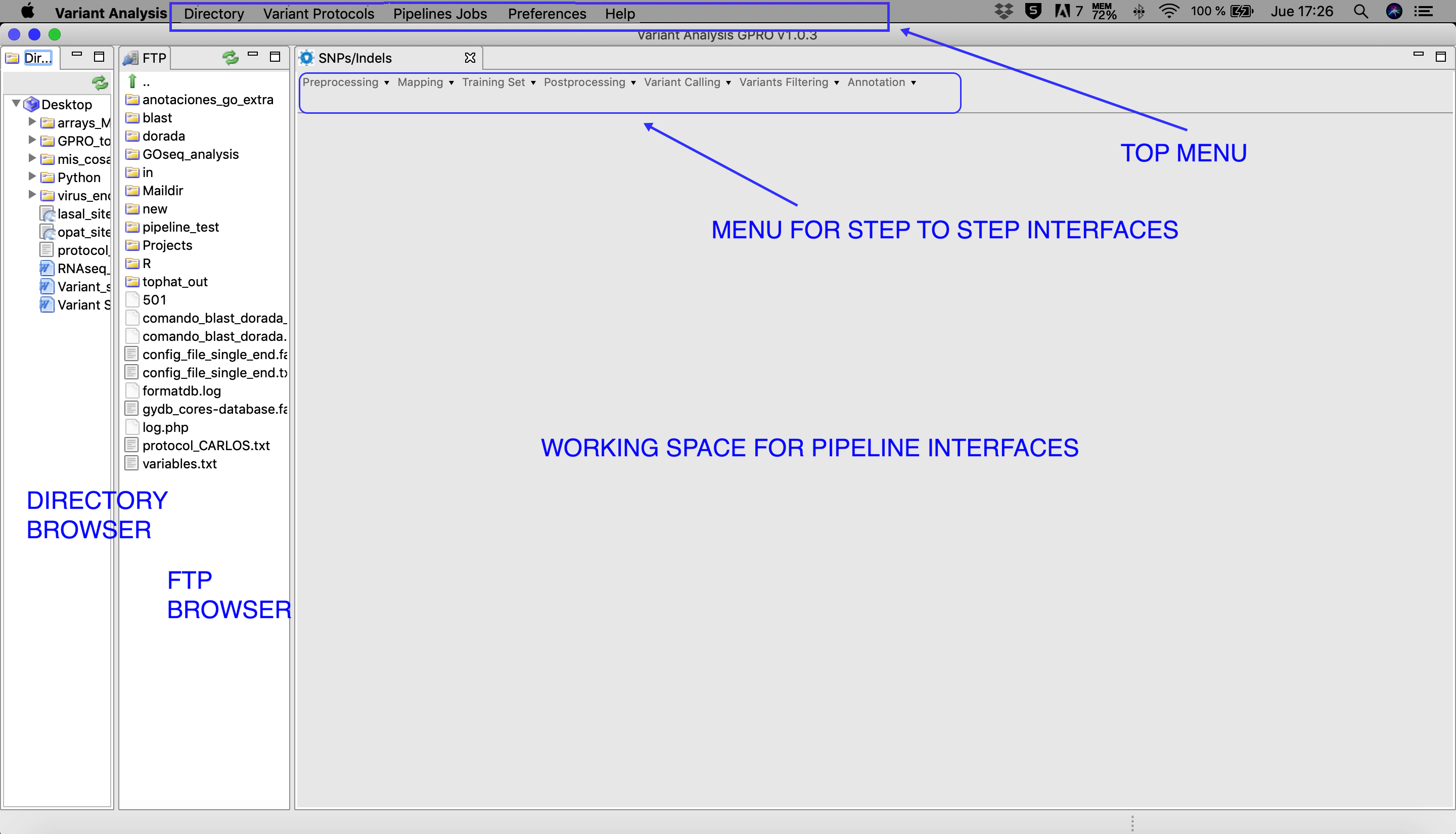
Figure 3: Main layout of VariantSeq. Both the Directory and FTP Browser windows can be resized or masked by clicking on the window icons in the top right corner of their respective windows. All files and folders contained in these browsers can be managed manually using the mouse. Please keep in mind that the window view shown in this manual will change depending on the operative system used.
[Directory → Select directory folder ] : select the Directory Browser.[Directory → Show ] : view the Directory Browser.[Directory → Hide ] : hide the Directory Browser.[Variant protocols -> Pipeline Mode → Experiment Configuration ] : Configuration view to set up your VariantSeq experiment and launch a VariantSeq pipeline.[Variant Protocols -> Pipeline Jobs → Step-by-Step → SNP/Indels Protocol ] : Jobs view for SNP/Indels Protocol in manual mode.[Pipeline Jobs → Jobs Tracking System ] : For jobs tracking.[Pipeline Jobs → FTP Transfers ] : screen for tracking the jobs of the sFTP protocol. [Preferences → Pipeline Connections Settings ] : setup your user credentials for accessing the server.[Help → Manual ] : Link to this manual.[Help → About VariantSeq ] : Other technical details and copyrigth of VariantSeq.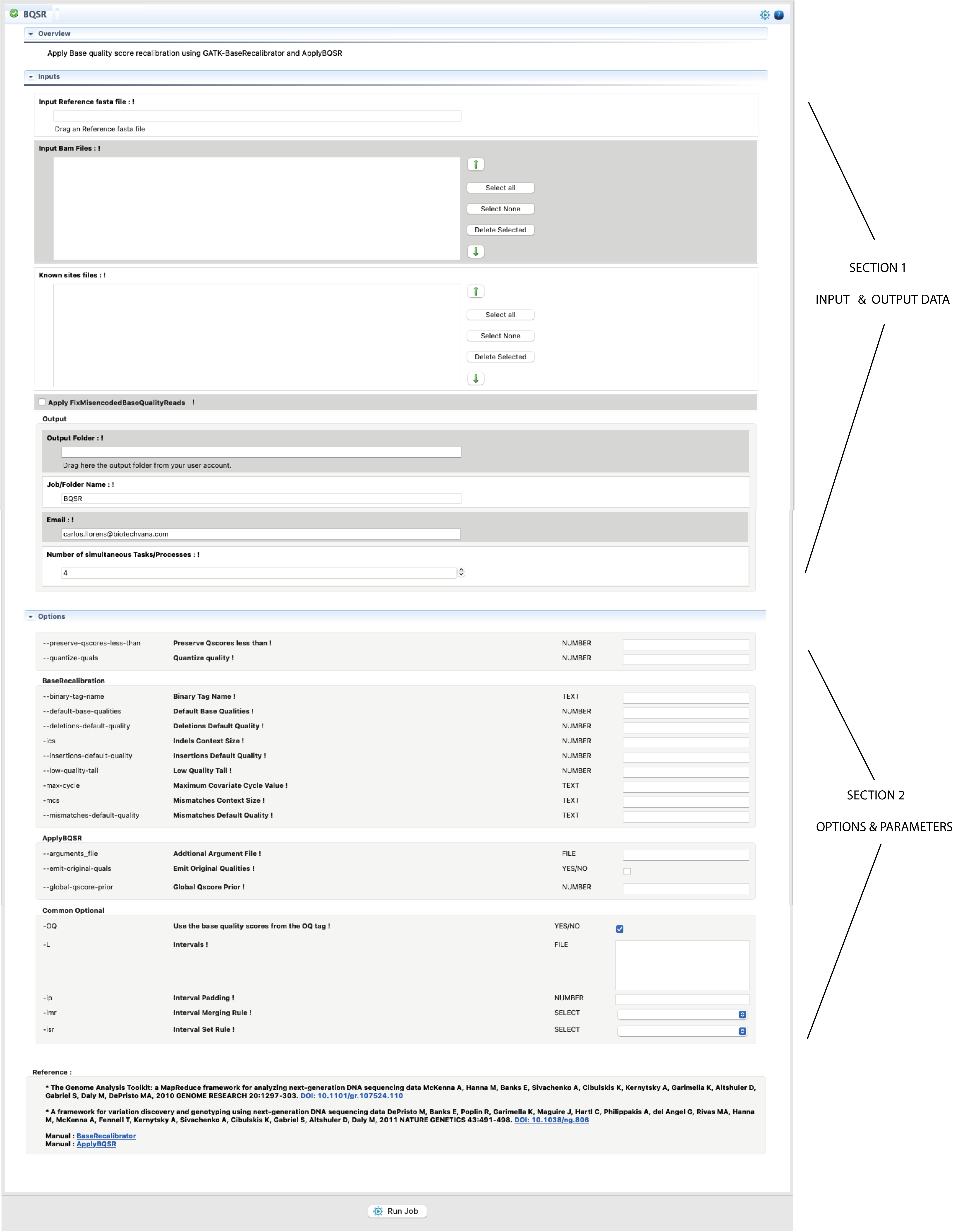
Figure 4: Example of interface for a CLI tool provided by the “VariantSeq” Step-By-Step mode. The figure shows the interface for the command BQSR of GATK.
The procedure is illustrated in Fig.4. Basically, you only need to select with the mouse the input file/s in FTP browser and drag it/them to their respective fields of the interface. The same can be done for the folder/s where you would like to have declared the output. Next, and if any, fill all other mandatory field(s) in the input/output section. If an input field is invalid or missing, you will get an error icon ![]() beside the field (to see the error message you will need to hover the mouse over that icon). Next, configure the form for options and parameters available in the second section. If you set a parameter out of the possible range, you will receive a warning icon
beside the field (to see the error message you will need to hover the mouse over that icon). Next, configure the form for options and parameters available in the second section. If you set a parameter out of the possible range, you will receive a warning icon ![]() . If you need more information about any input or parameter, click on the exclamation mark ! in front of the input field name.
. If you need more information about any input or parameter, click on the exclamation mark ! in front of the input field name.
Filling the section of options and parameters is not mandatory. If you run the program without inputting any parameters and/or condition, VariantSeq will execute this program using the default conditions for this program.
Once the job input fields and the parameters have been fulfilled and/or configured, you only need to click to start button at the end the interface form to run the Job. If the job has been successfully launched, you will get a confirmation message. Otherwise revise again all input and outputs upload and the options if any.







