To make use of the different protocols of VariantSeq for calling and annotation of SNPs and Indels, go to:
[Variant Protocols → Step-by-Step → SNP/Indels]
A new submenu will appear (Fig. 5) in the workspace listing the distinct steps of the variant analysis (Preprocessing, mapping, Trainings Sets, Postprocessing, Variant Calling, and Variant Filtering).
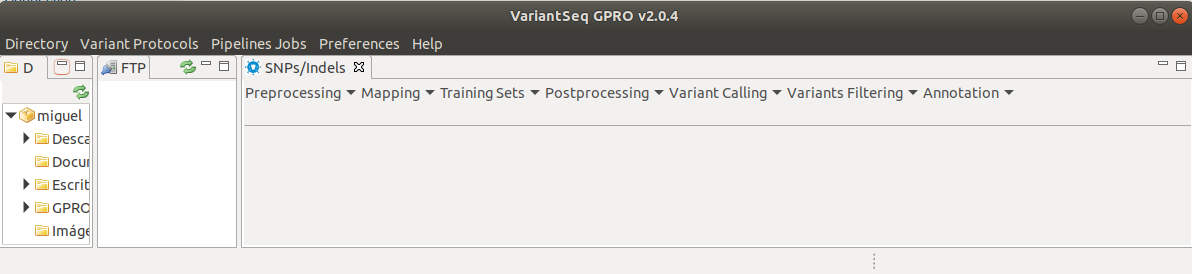
Figure 5: Submenu to follow the SNP/Indell calling protocol In Step-by-step mode.
Raw data preprocessing is necessary to prepare the fastq libraries for mapping and this job involves several steps in which the raw reads are trimmed, cleaned or modified to remove adapter remains, sequencing artifacts, contaminations and/or low quality sequences. The “preprocessing” drop-down submenu (Fig. 6) provides access to the tools for quality analysis, demultiplex, and other sequence trimming & cleaning tools including those for adapter removal or filtering out low quality sequences. The following sections provide a brief description of each preprocessing tool.
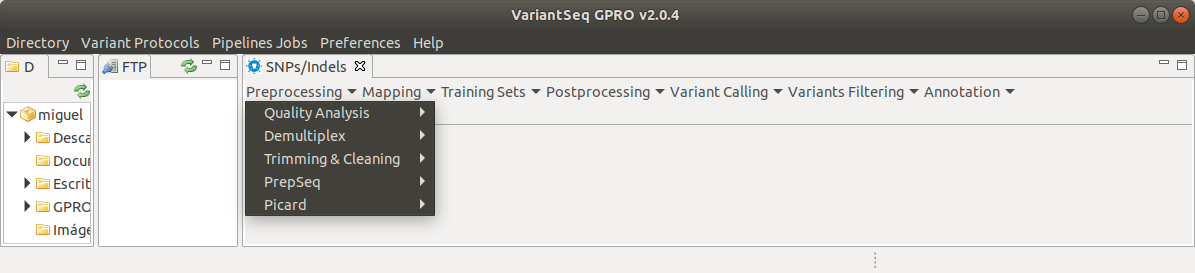
Figure 6: Preprocessing drop down submenu listing the preprocessing options enabled in the VariantSeq application.
- Quality Analysis: FastQC
FastQC (Andrews 2016) provides a simple way to perform quality checks on raw sequence data. It provides a modular set of analyses that can indicate whether your data contain potential artifacts that require “cleaning” before beginning any analyses. Upon performing a FastQC quality check, you will obtain a complete sequence quality report provide hints on what form of filtering and processing your sample requires if any. For example, overrepresented sequences that correspond to the adapters used during sequencing may need to be removed from your fastq files. This will be shown in the “overrepresented sequences” section of the FastQC report. Further details on FastQC can be found in the FastQC manual at https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
To run FastQC, go to [ Preprocessing → Quality Analysis → FastQC ] and follow Fig. 7.
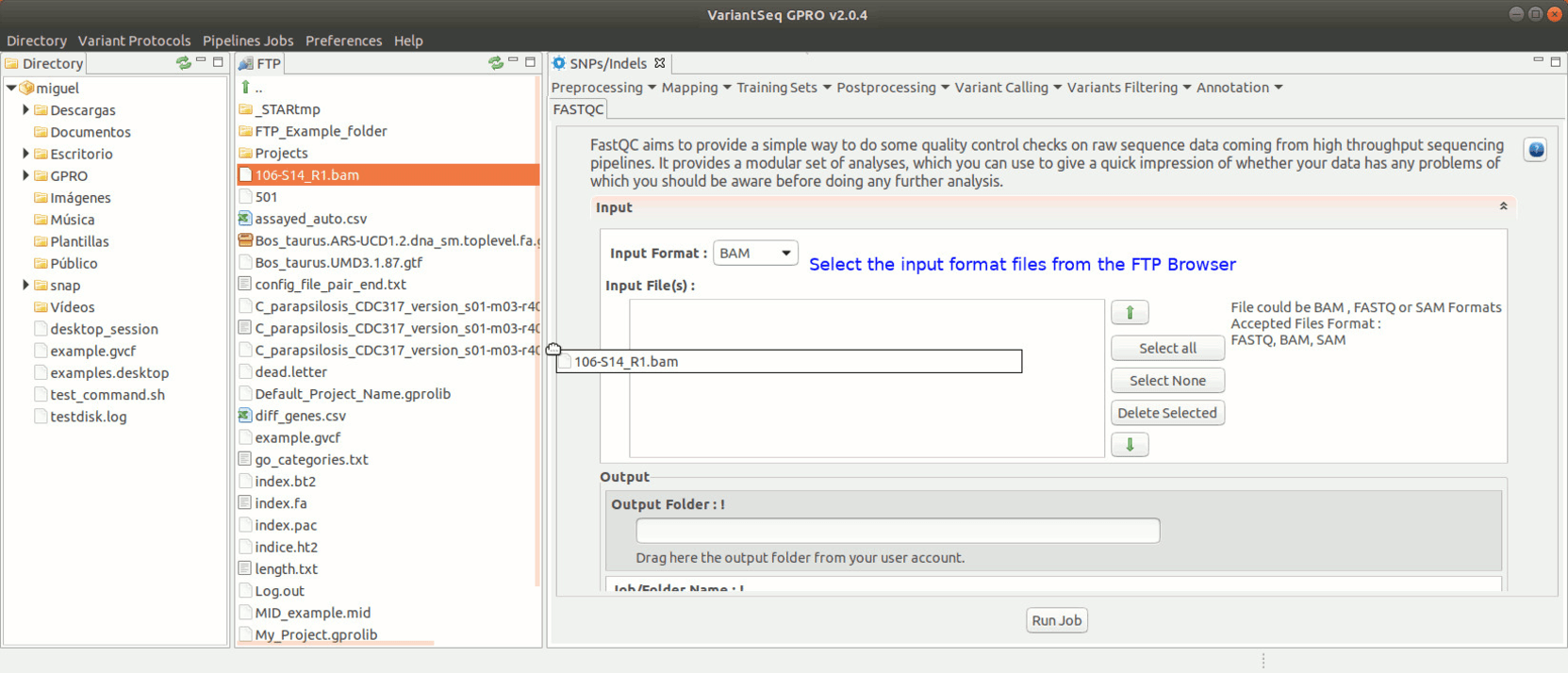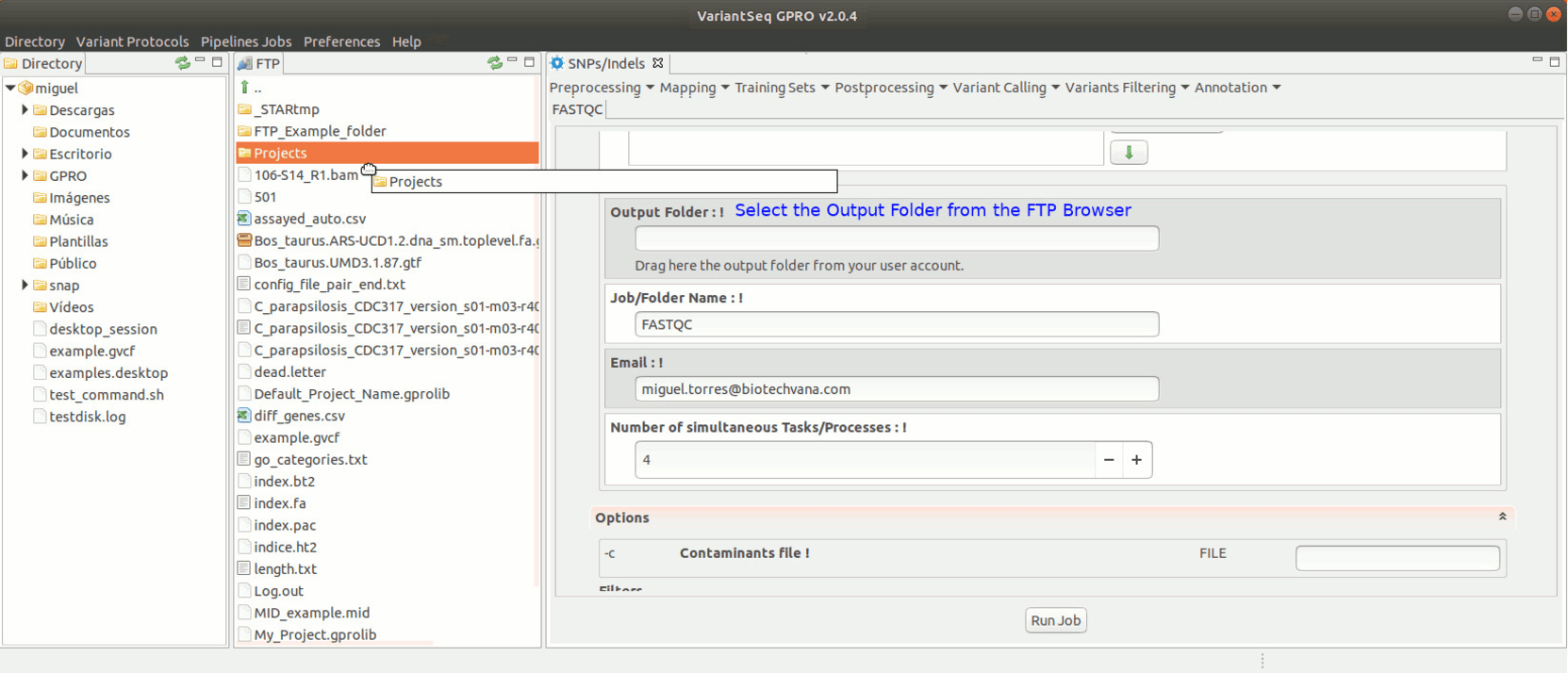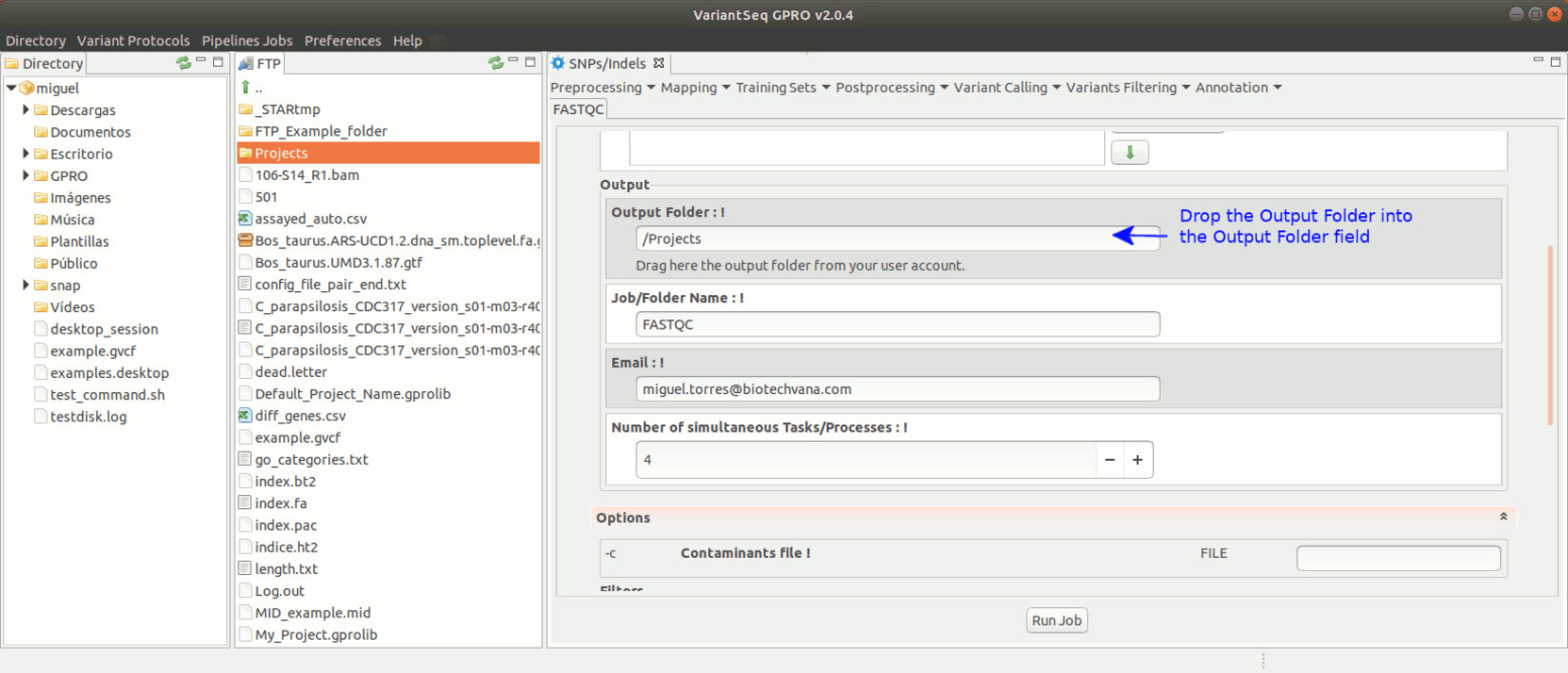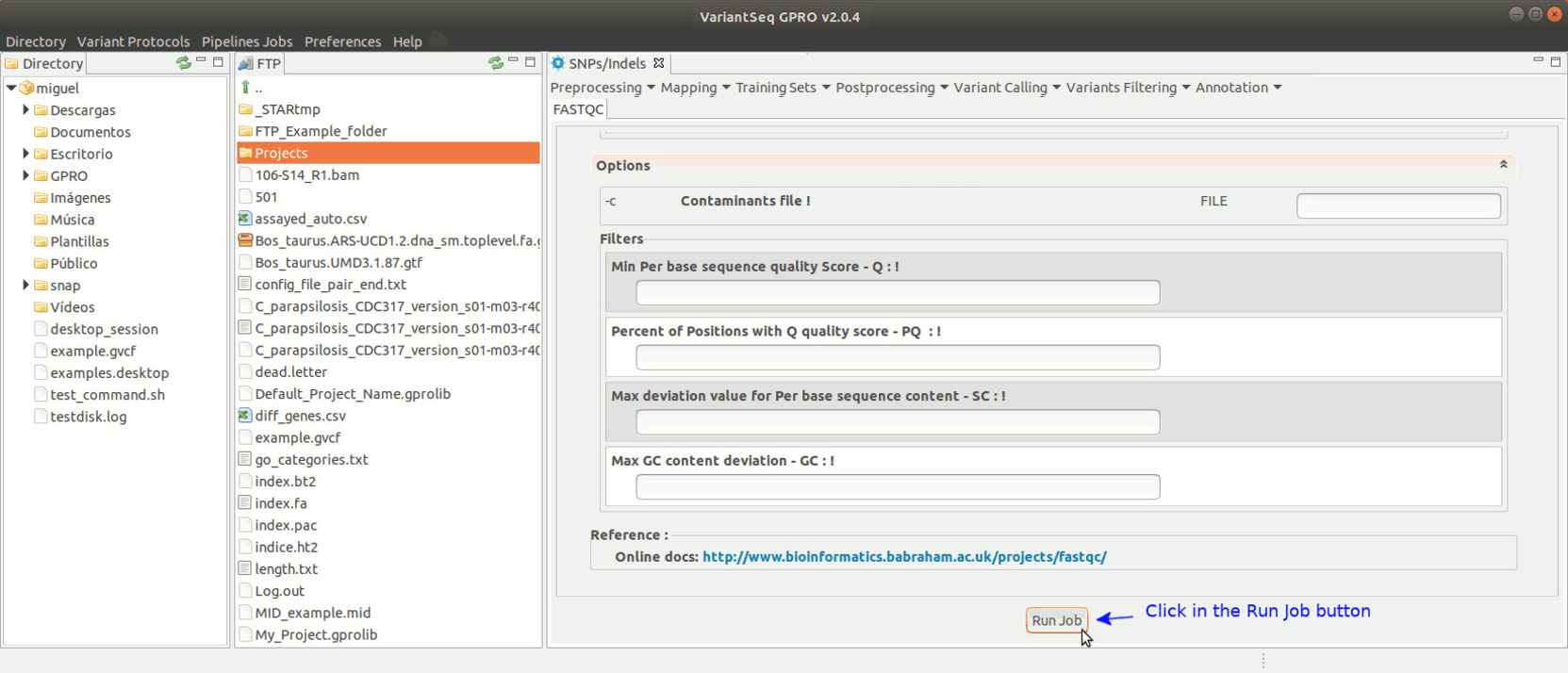
Figure 7: Using the GPRO interface for FastQC.
- Demultiplex: FastqMidCleaner
FastqMidCleaner sorts and splits sequencing reads from fastq files into separate files according to predefined molecular identifiers (MIDs).
To run FastqMidCleaner with VariantSeq go to [Preprocessing→Demultiplex → FastqMidCleaner ] and follow Fig.8.
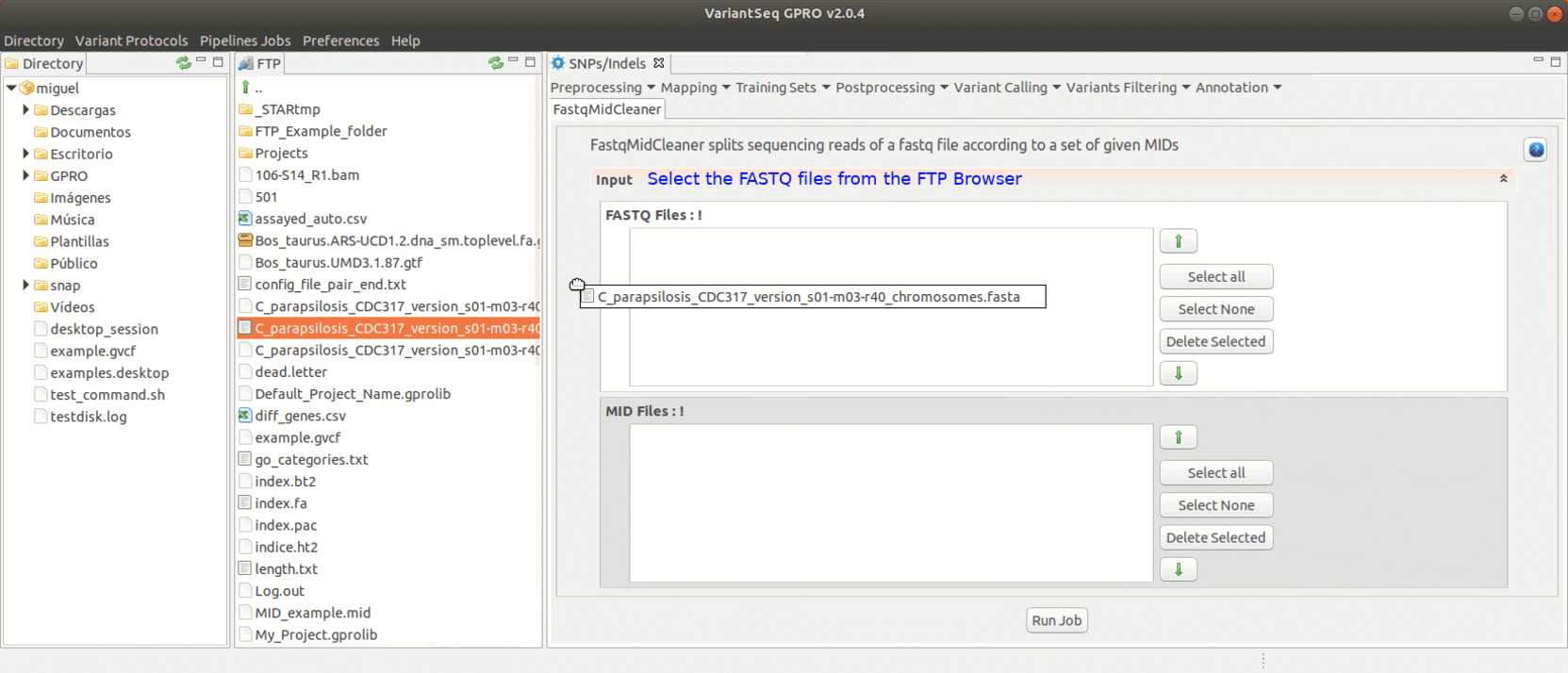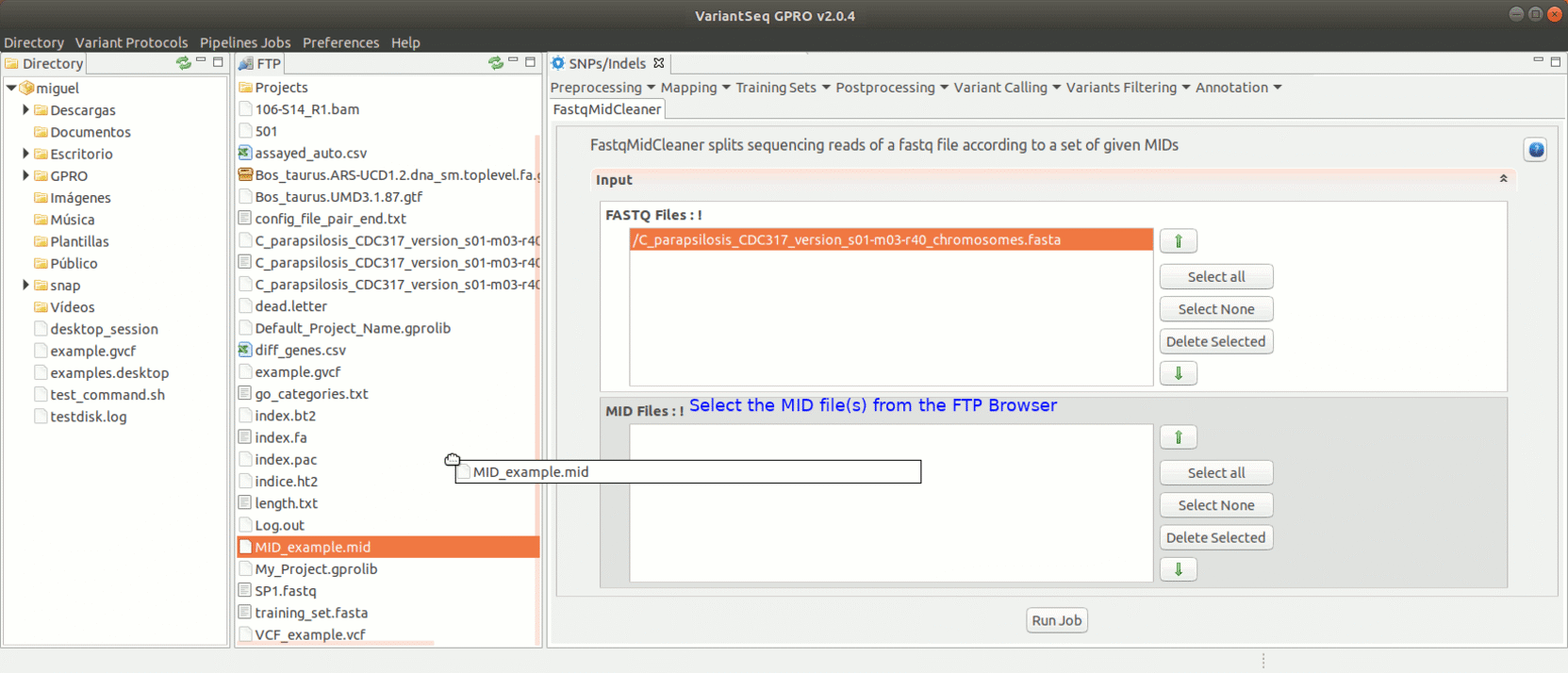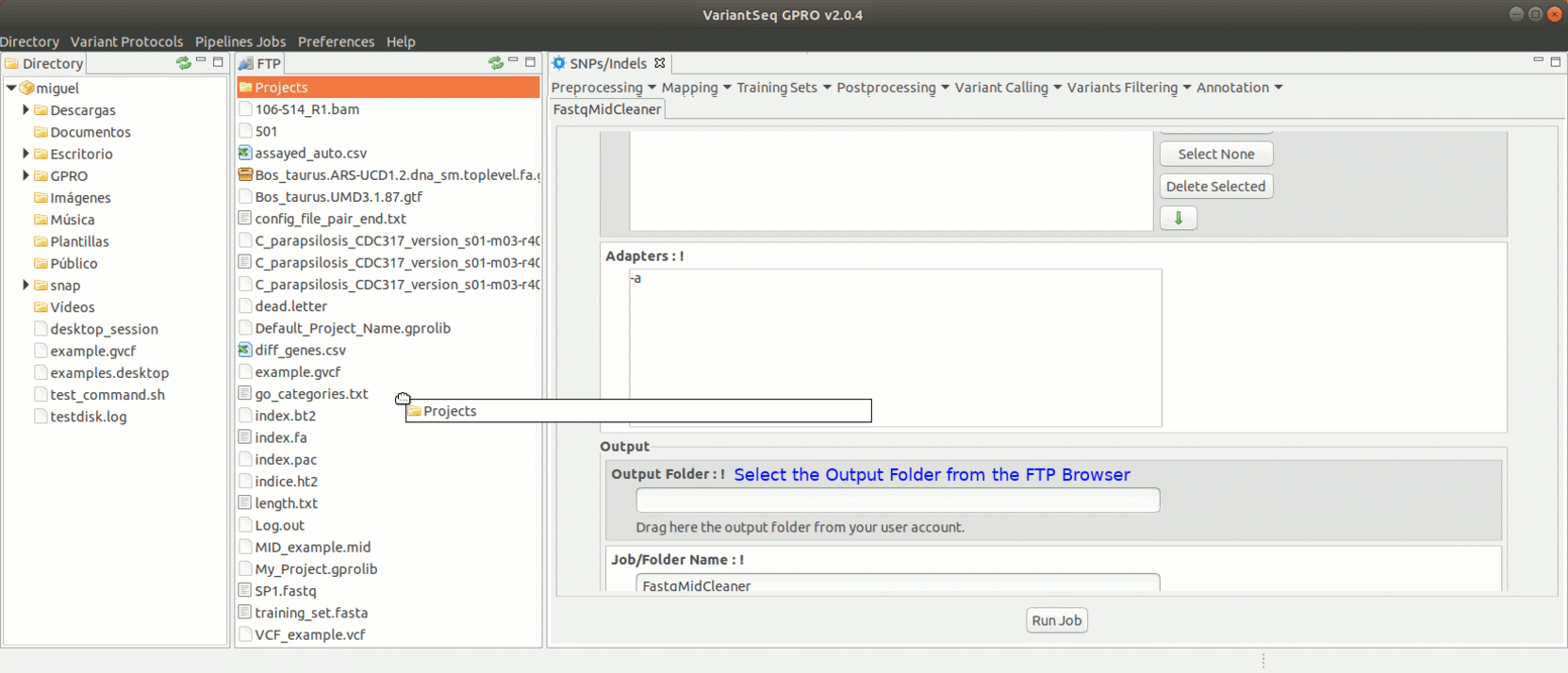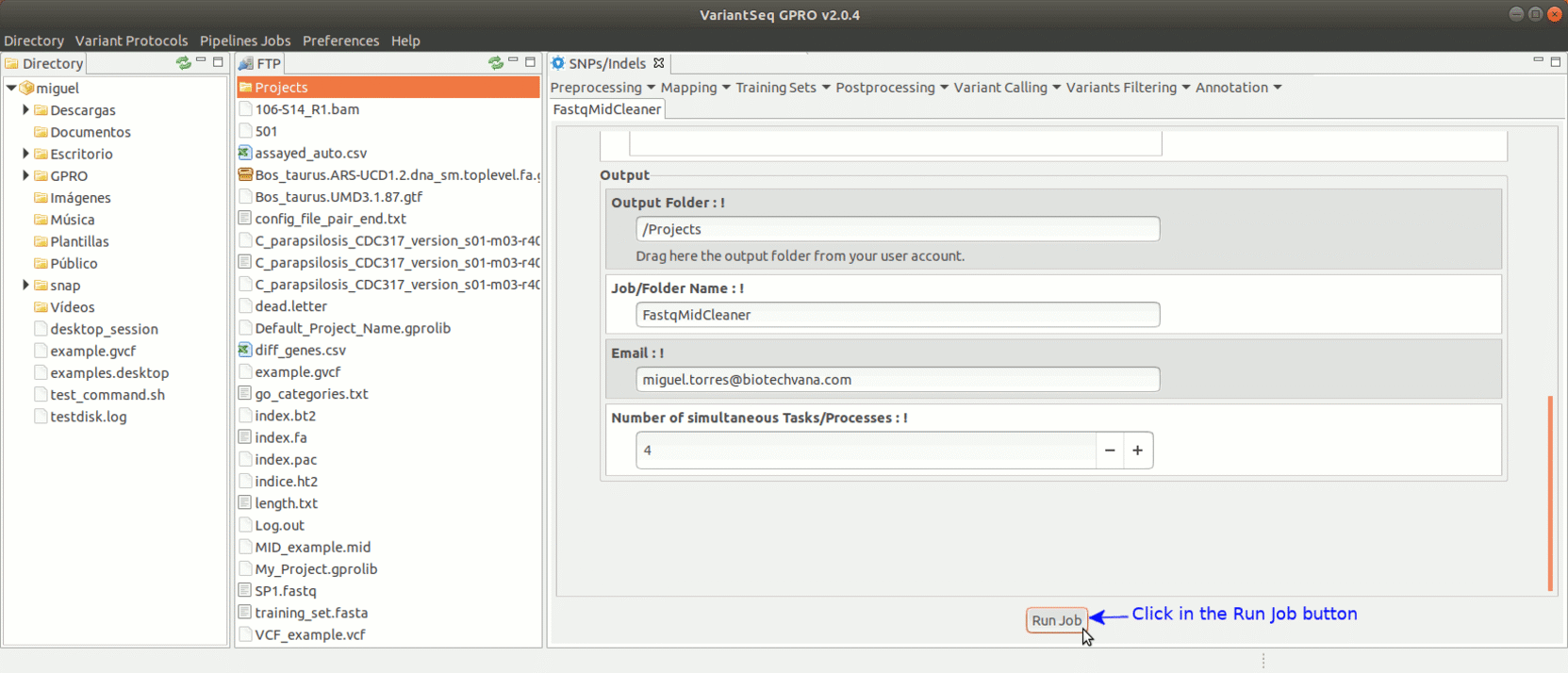
Figure 8: Using the GPRO interface for FastQMidCleaner.
Cutadapt (Martin 2011) finds and removes adapter sequences, primers, poly-A tails and other types of unwanted sequences from your sequencing reads. For more information on Cutadapt see the Cutadapt manual at https://cutadapt.readthedocs.io/en/stable/guide.html.
To run Cutadapt, go to [ Preprocessing→ Trimming & Cleaning → Cutadapt ] and follow Fig. 9.
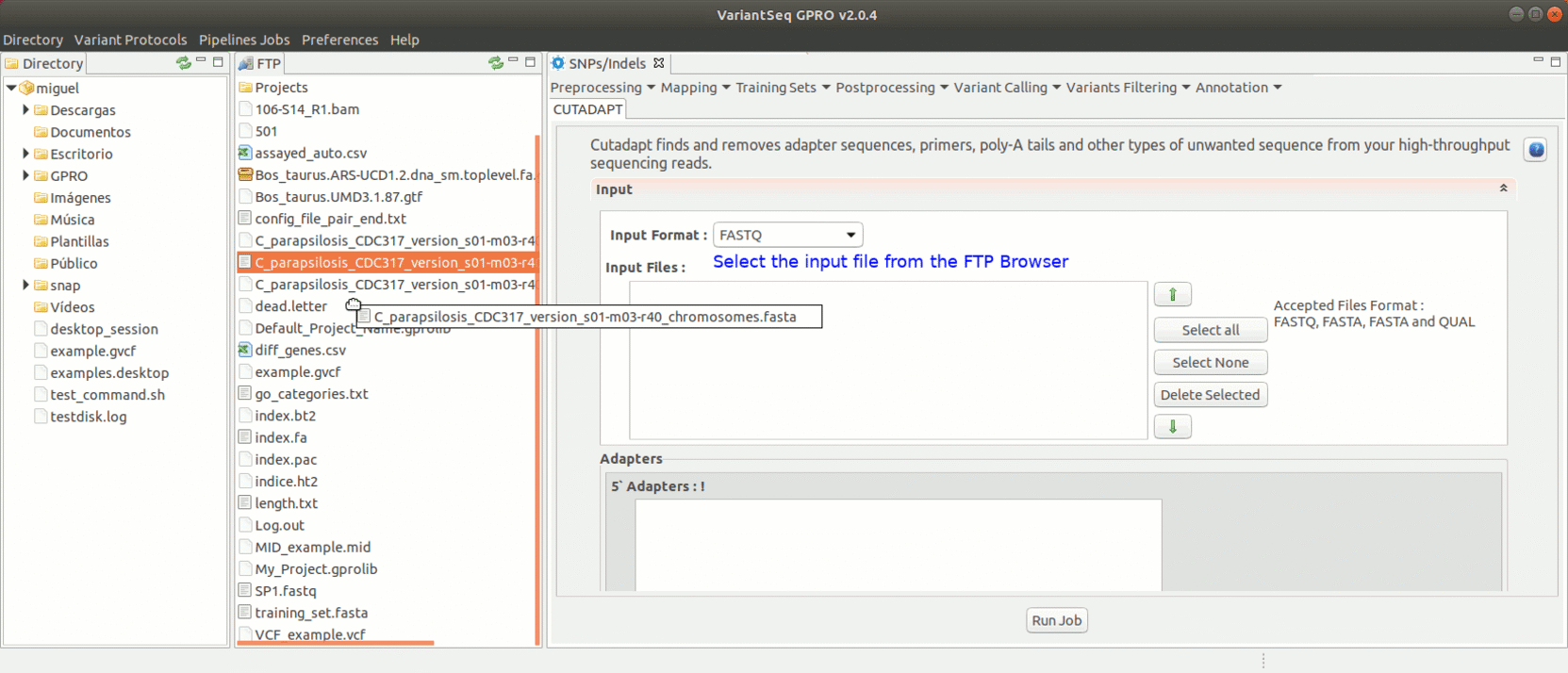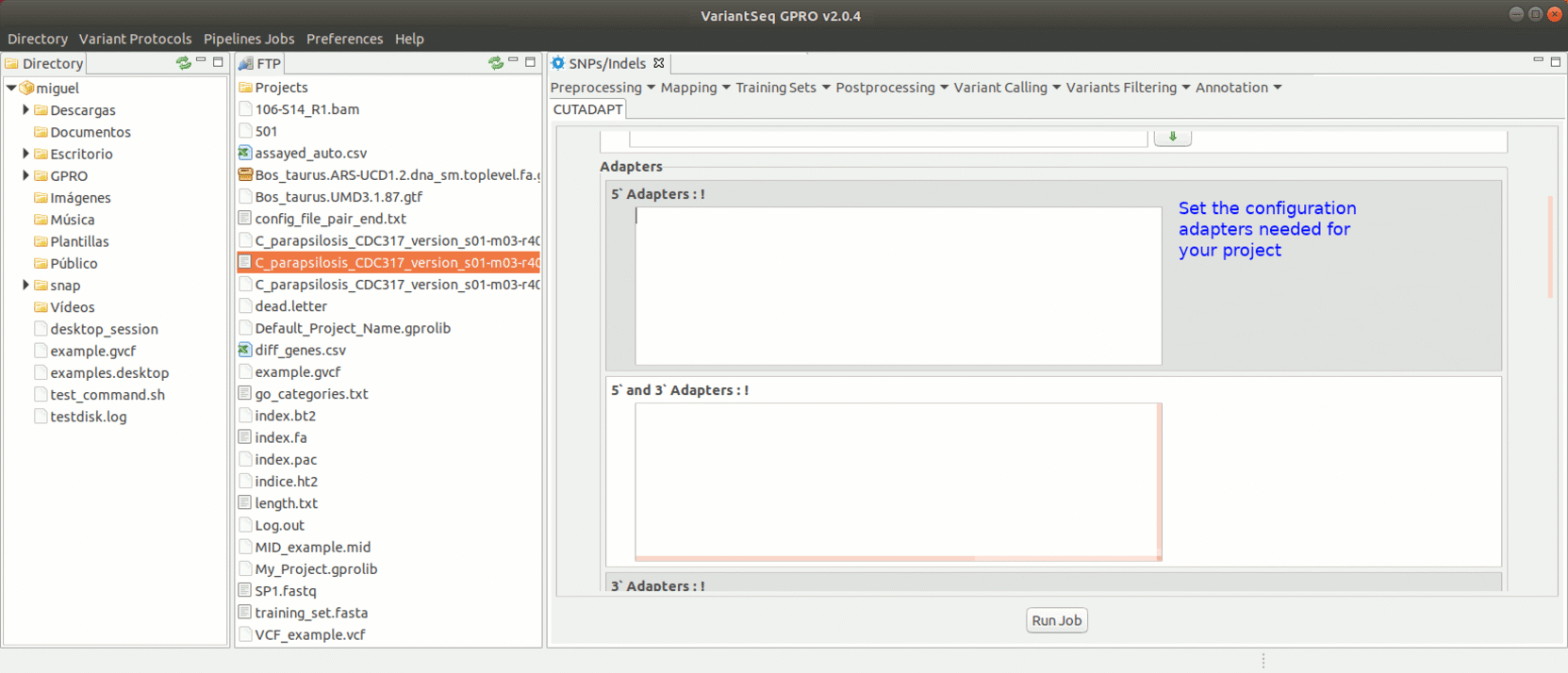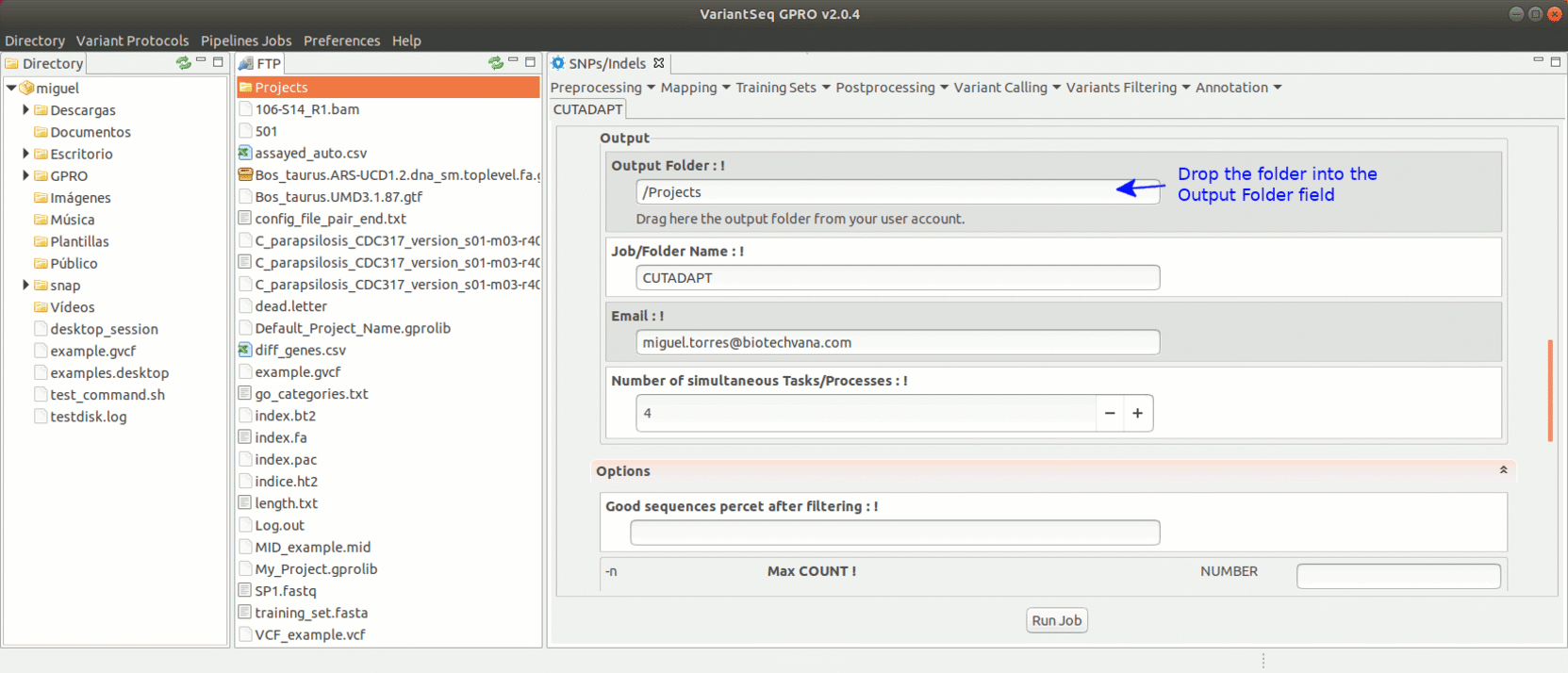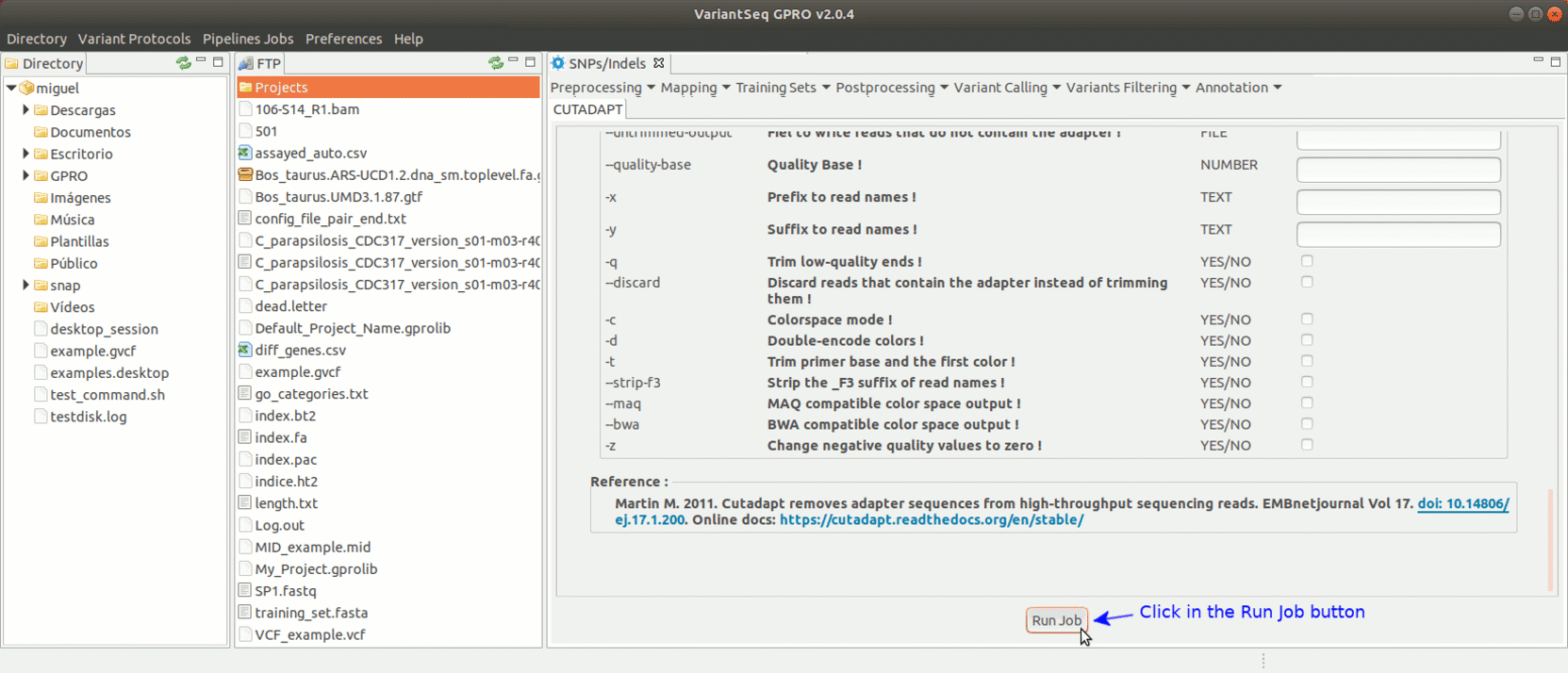
Figure 9: Using the GPRO interface for Cutadapt.
- Trimming & Cleaning: Prinseq
Prinseq (Schmieder and Edwards 2011) can filter, reformat, or trim your sequencing reads. For further information, see the Prinseq manual at http://prinseq.sourceforge.net.
To run Prinseq, go to [Preprocessing→Trimming & Cleaning → prinseq ] and follow Fig. 10.
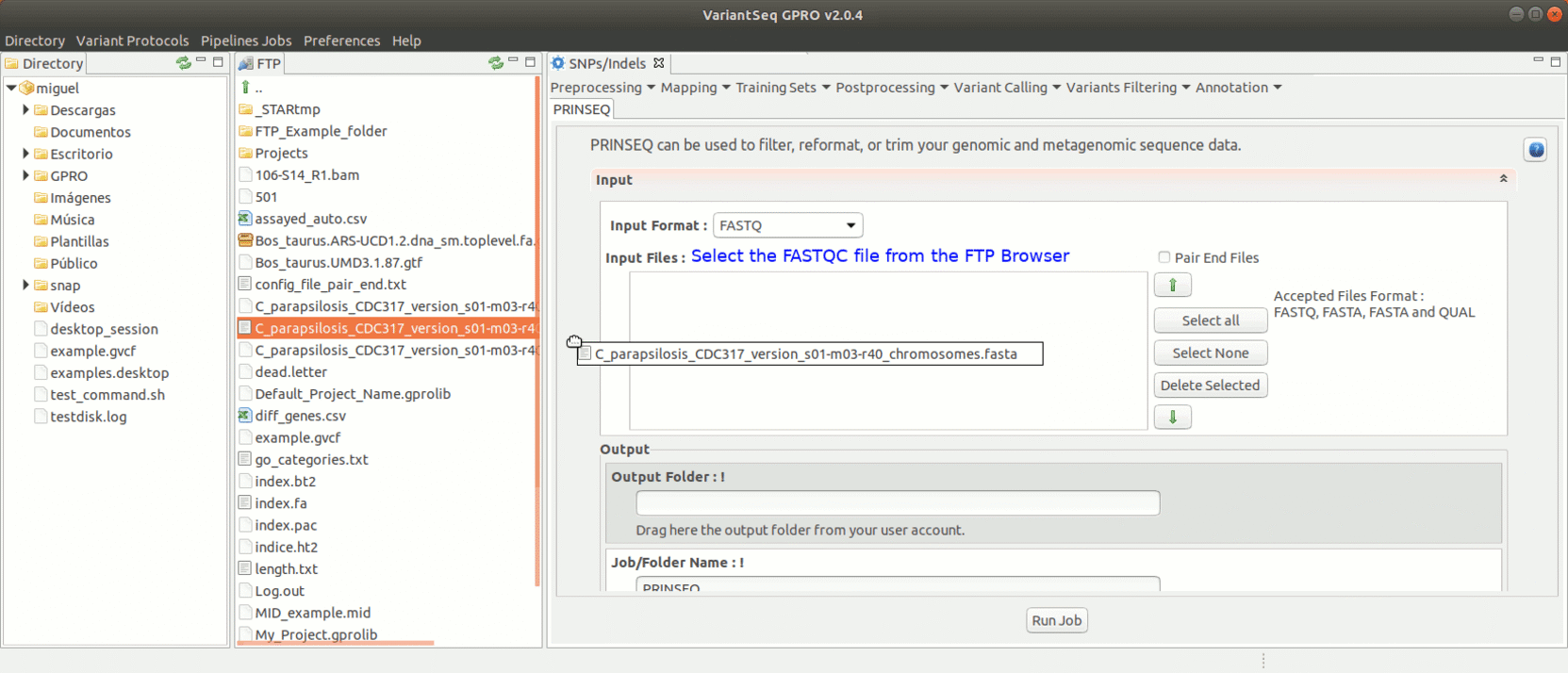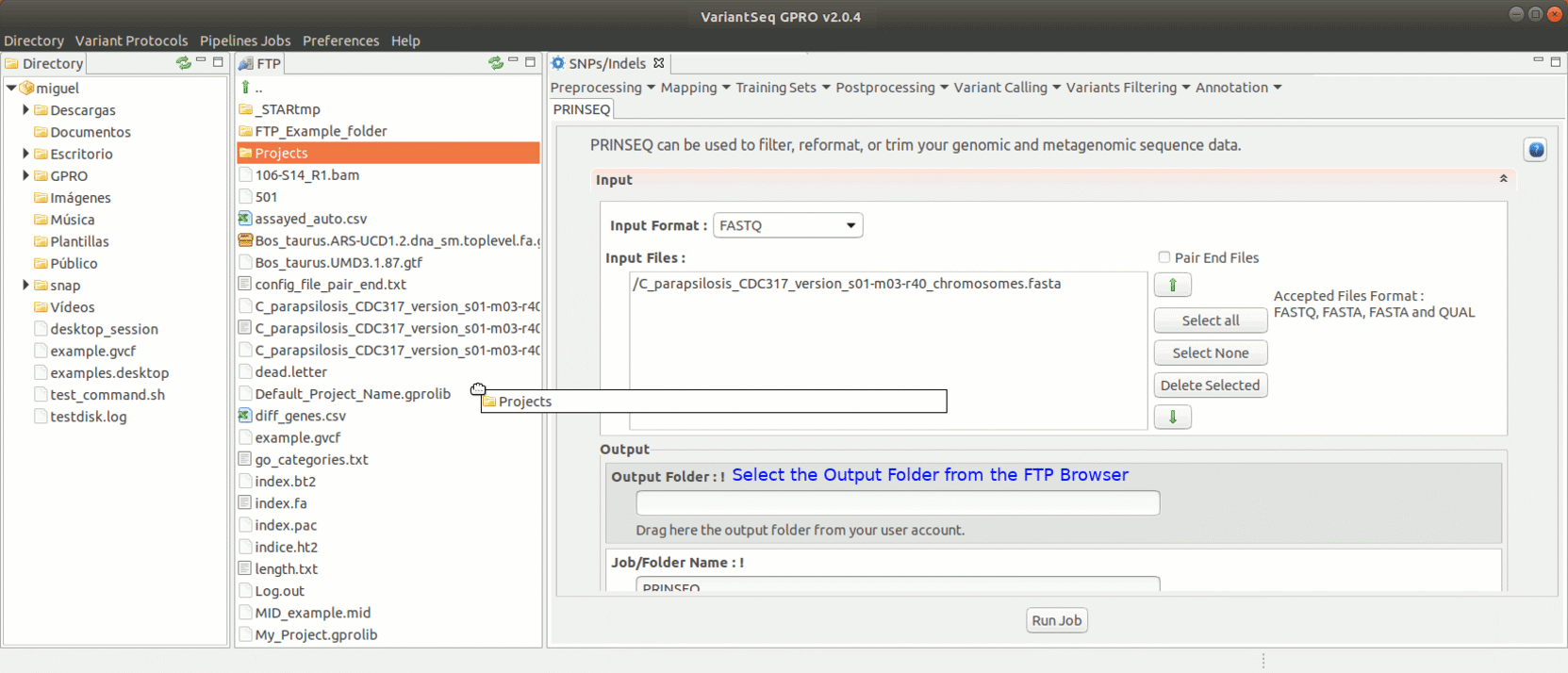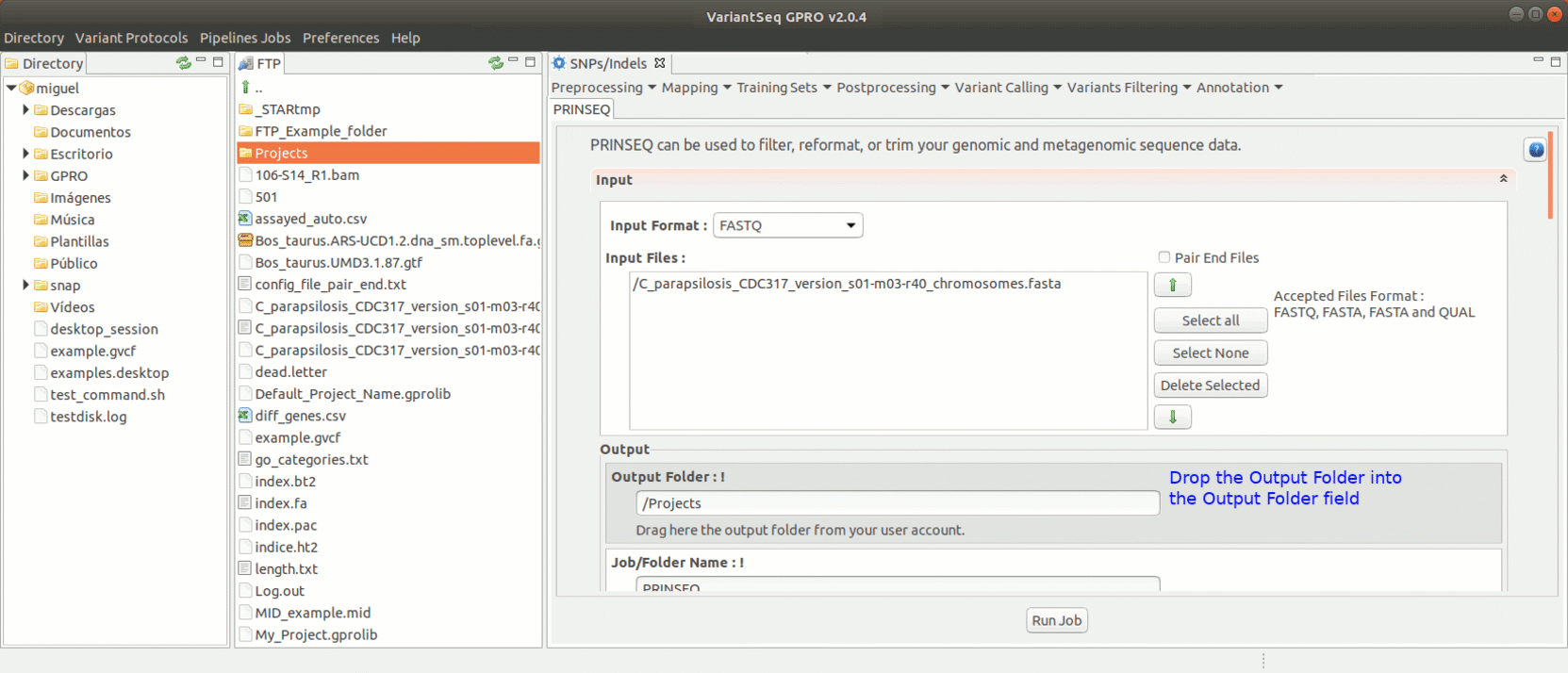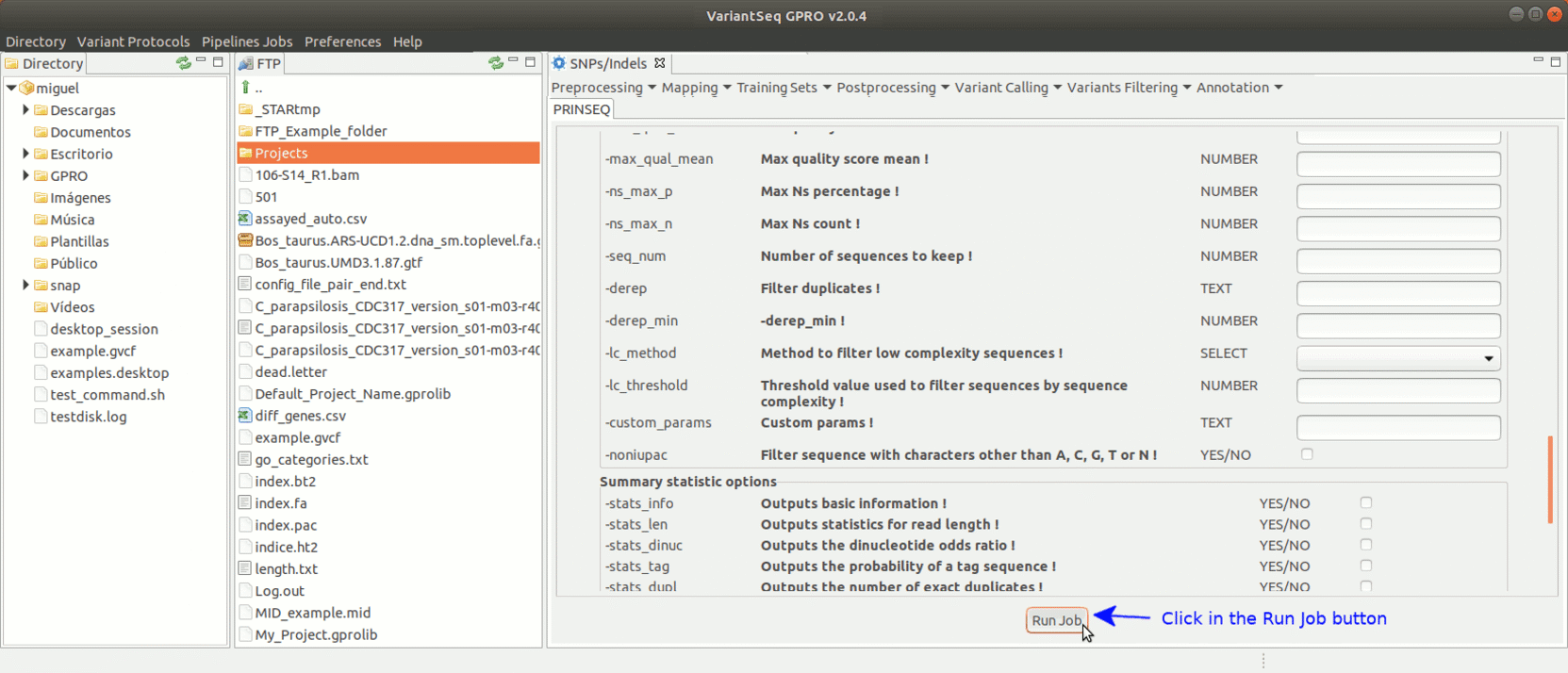
Figure 10: Using the GPRO interface for Prinseq.
- Trimming & Cleaning: Trimmomatic
Trimmomatic (Bolger et al. 2014) is trimming tool specific for paired-end and single-end reads obtained via Illumina’s NGS technology. For more information, see the trimmomatic manual at http://www.usadellab.org/cms/?page=trimmomatic.
To run Trimmomatic, go to [Preprocessing→Trimming & Cleaning → trimmomatic] and follow Fig. 11.
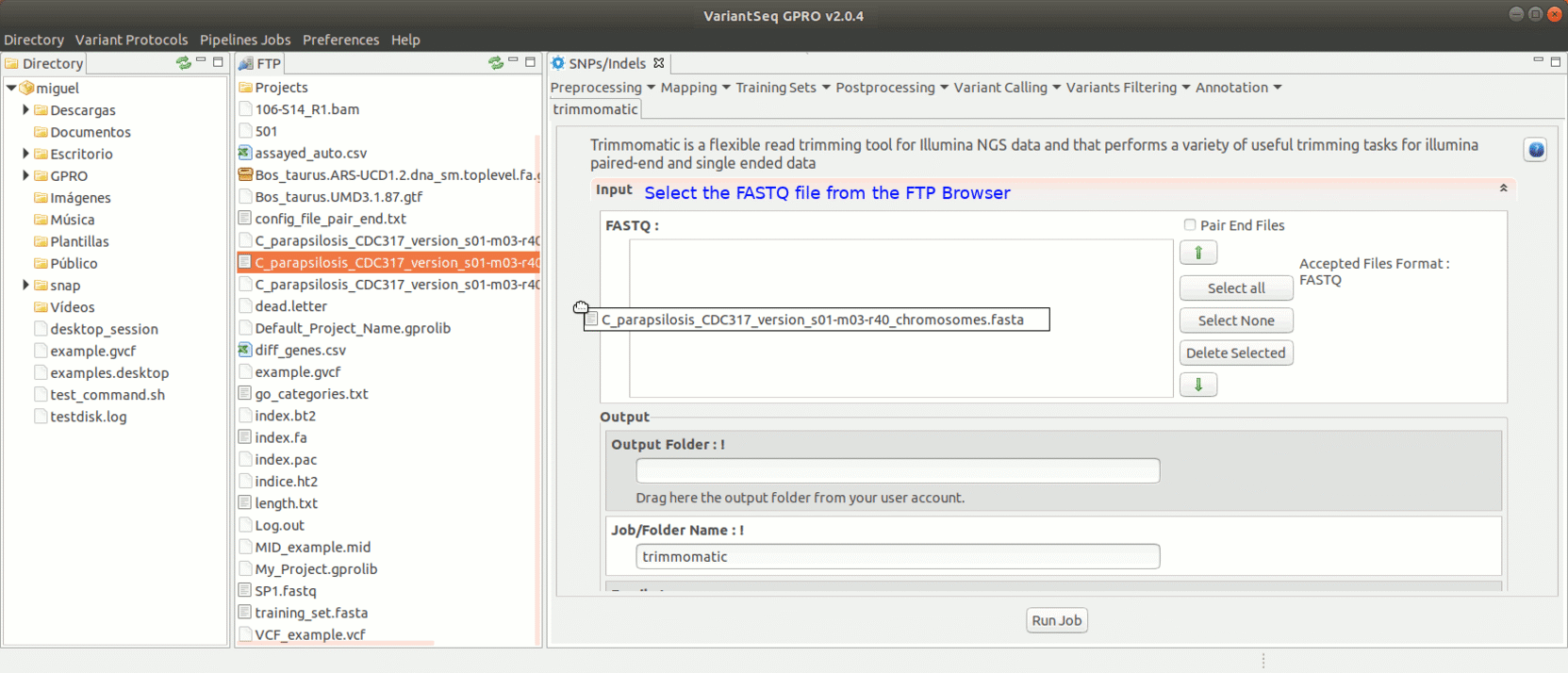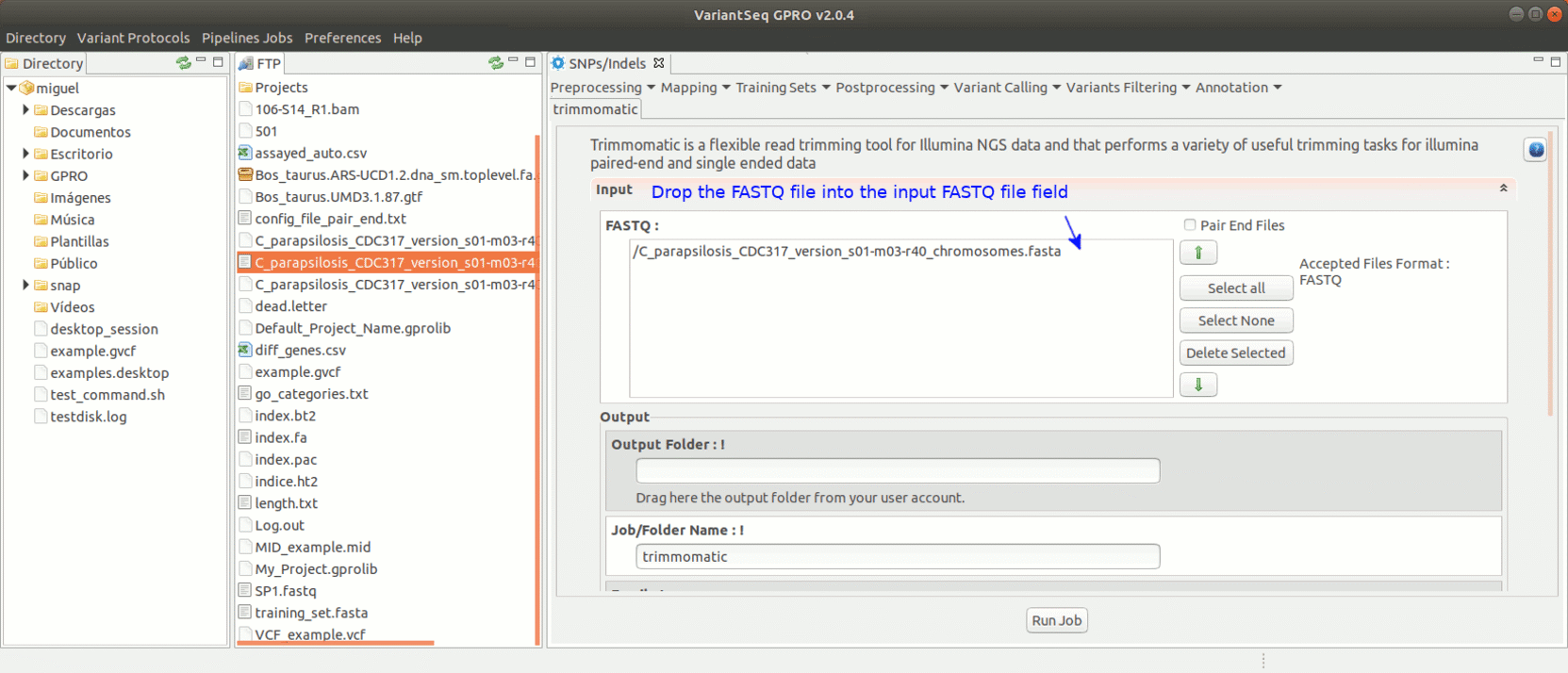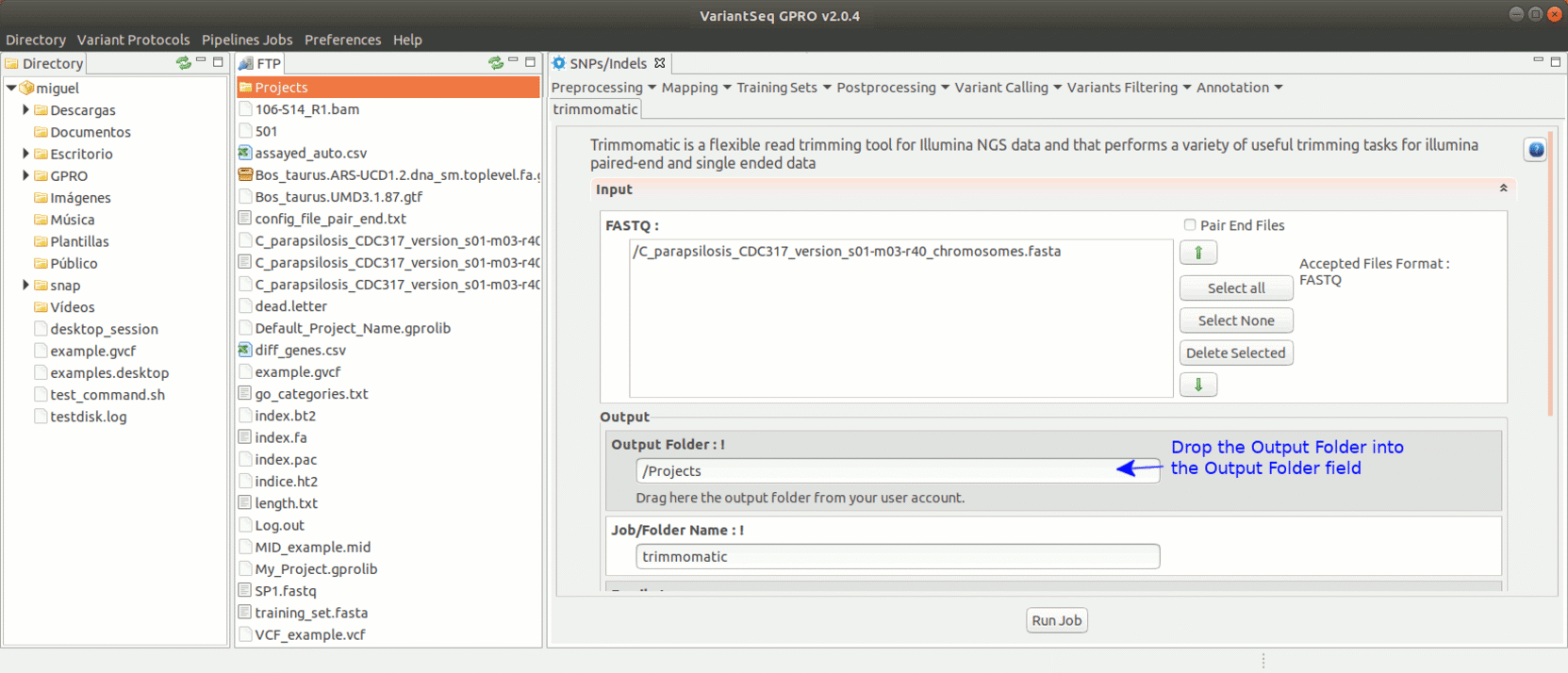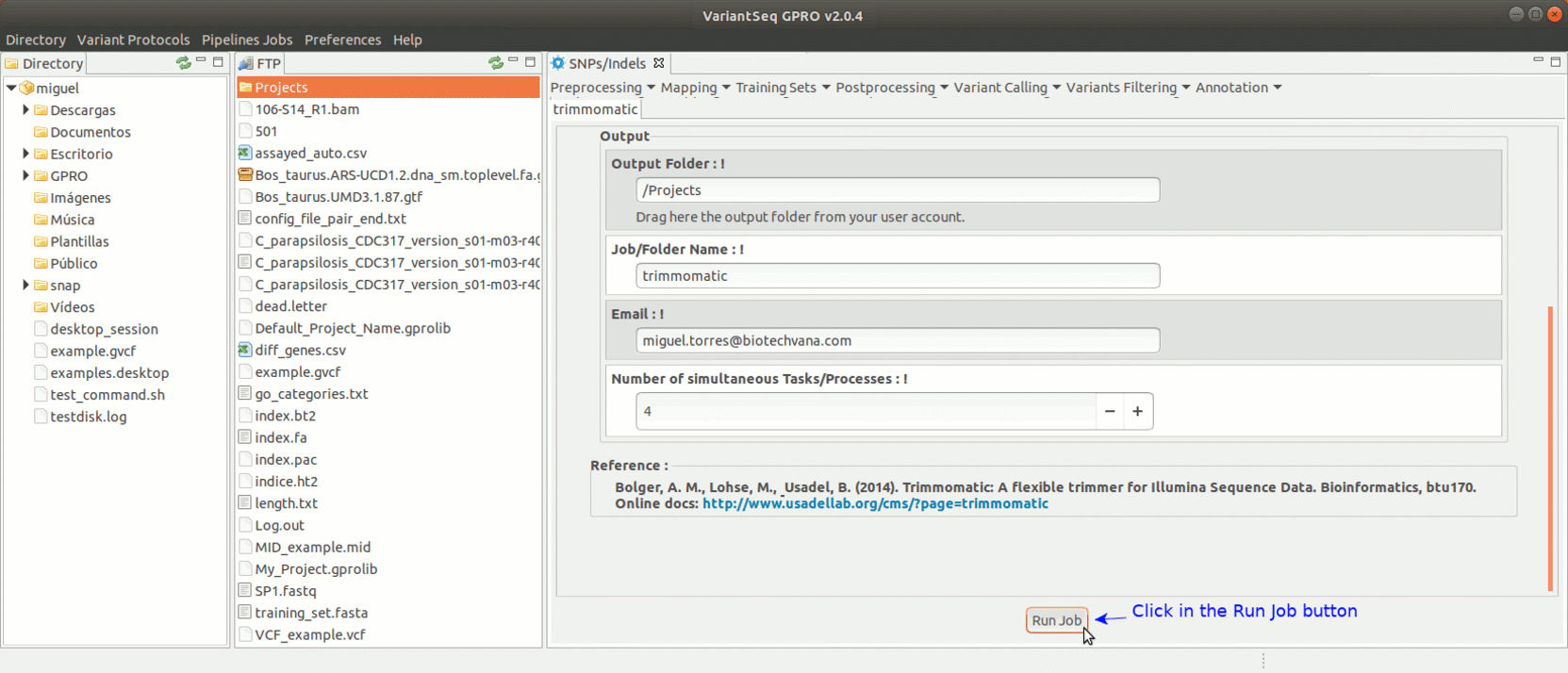
Figure 11: Using the GPRO interface for Trimmomatic.
- Trimming & Cleaning: FastxToolKit
FASTX-Toolkit (Hannon Lab 2016) is a set preprocessing tools for Fasta/Fastq files that include the following:
To run the FastxToolKit, go to [Preprocessing→ Trimming & Cleaning → Fastx-Toolkit] and follow Fig. 12.
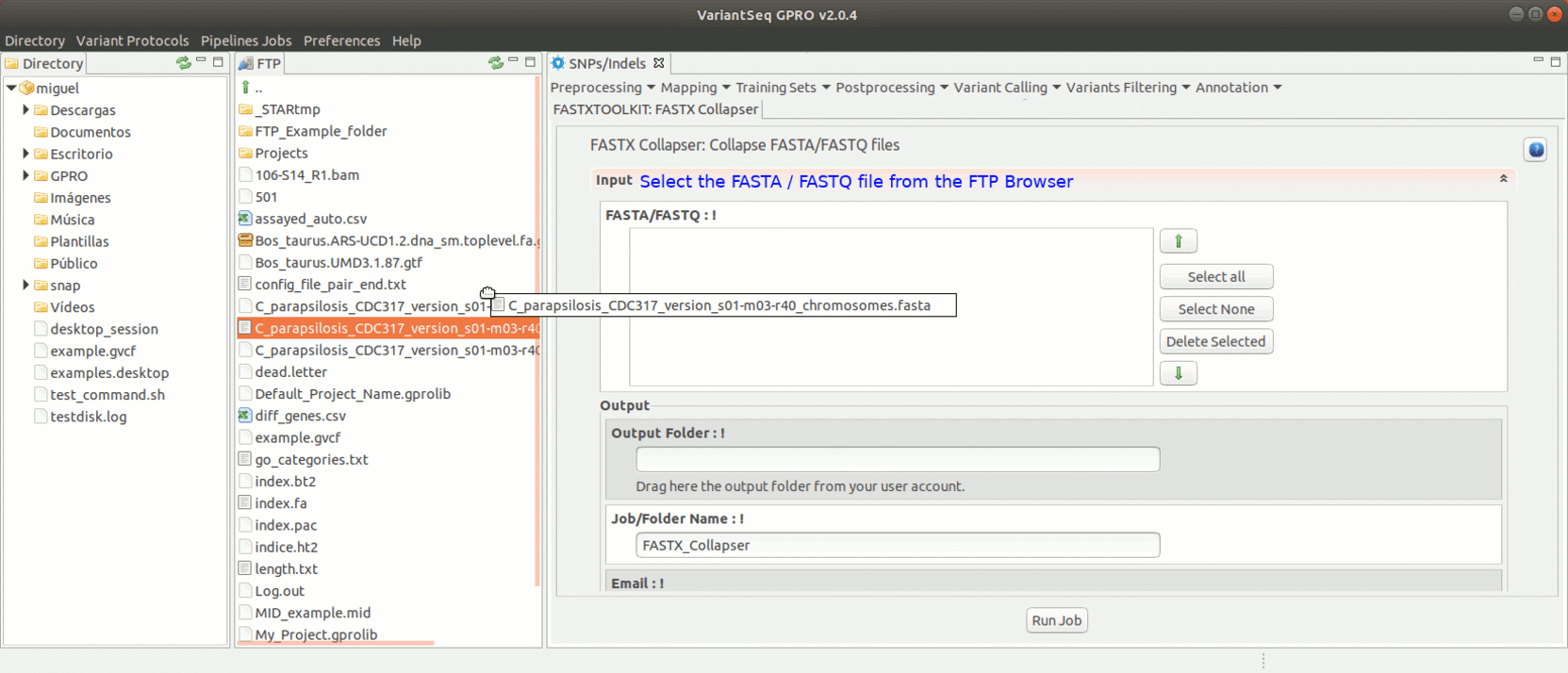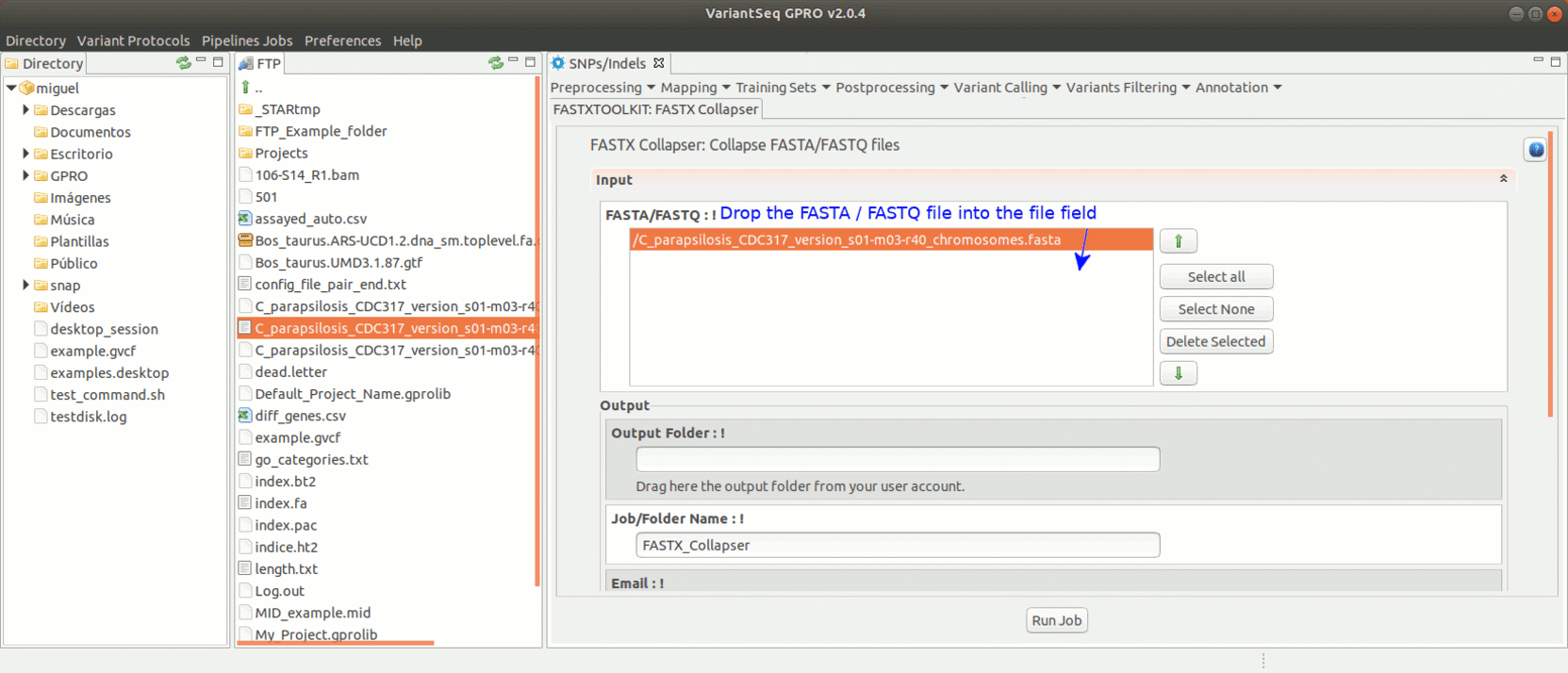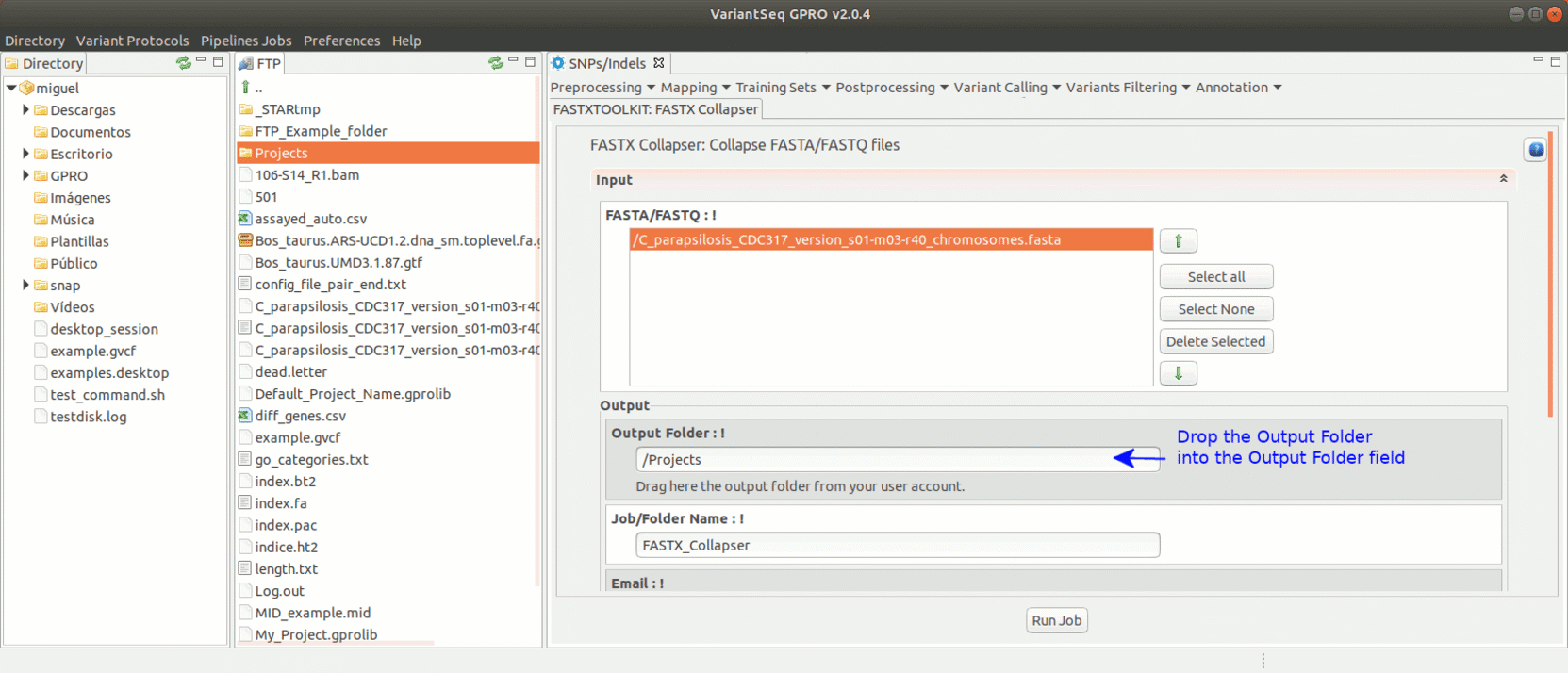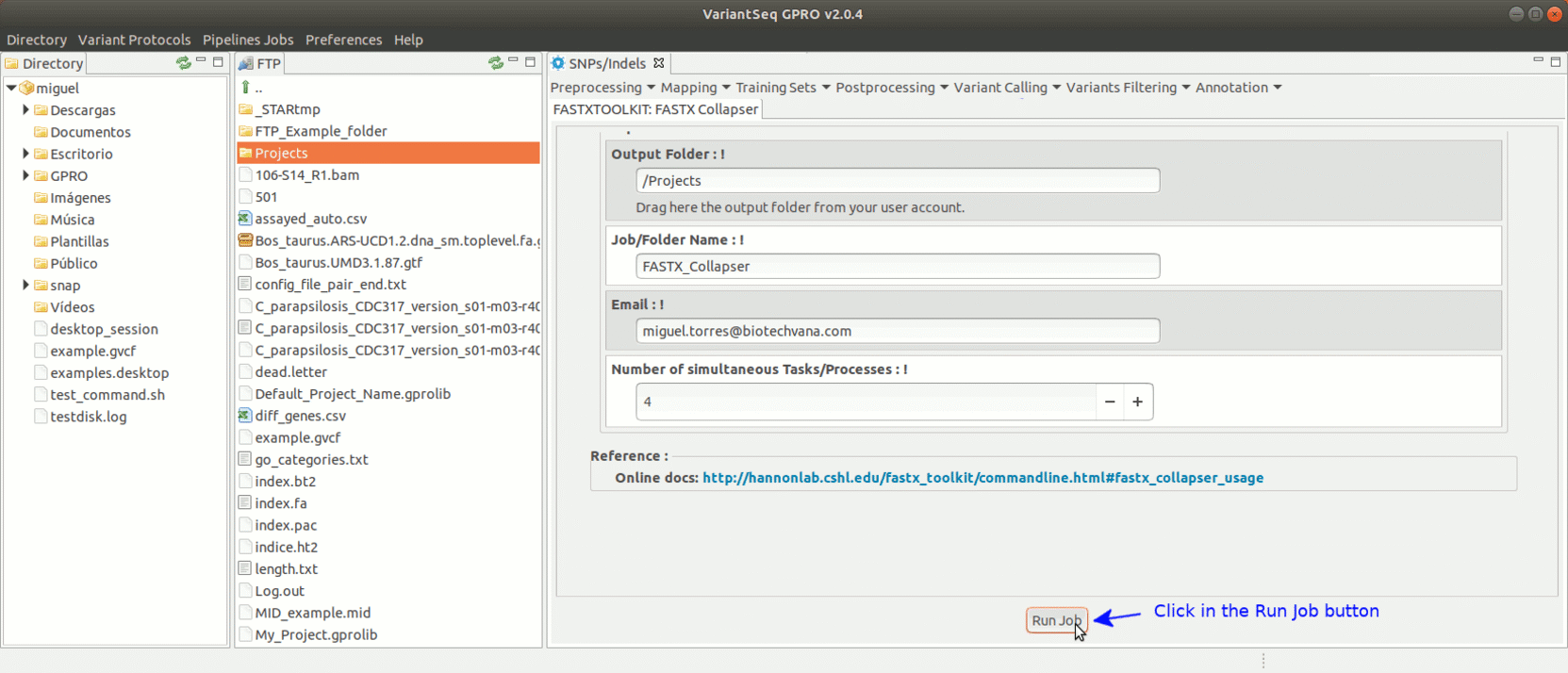
Figure 12: Example of GPRO interface for a tool (FASTX collapser) of Fastx-Toolkit.
- PrepSeq: FastqCollapser
FastqCollapser is used to remove duplicate reads from fastq files based on their sequence content.
To run FastqCollapser, go to [ Preprocessing→ PrepSeq → FastqCollapser ]
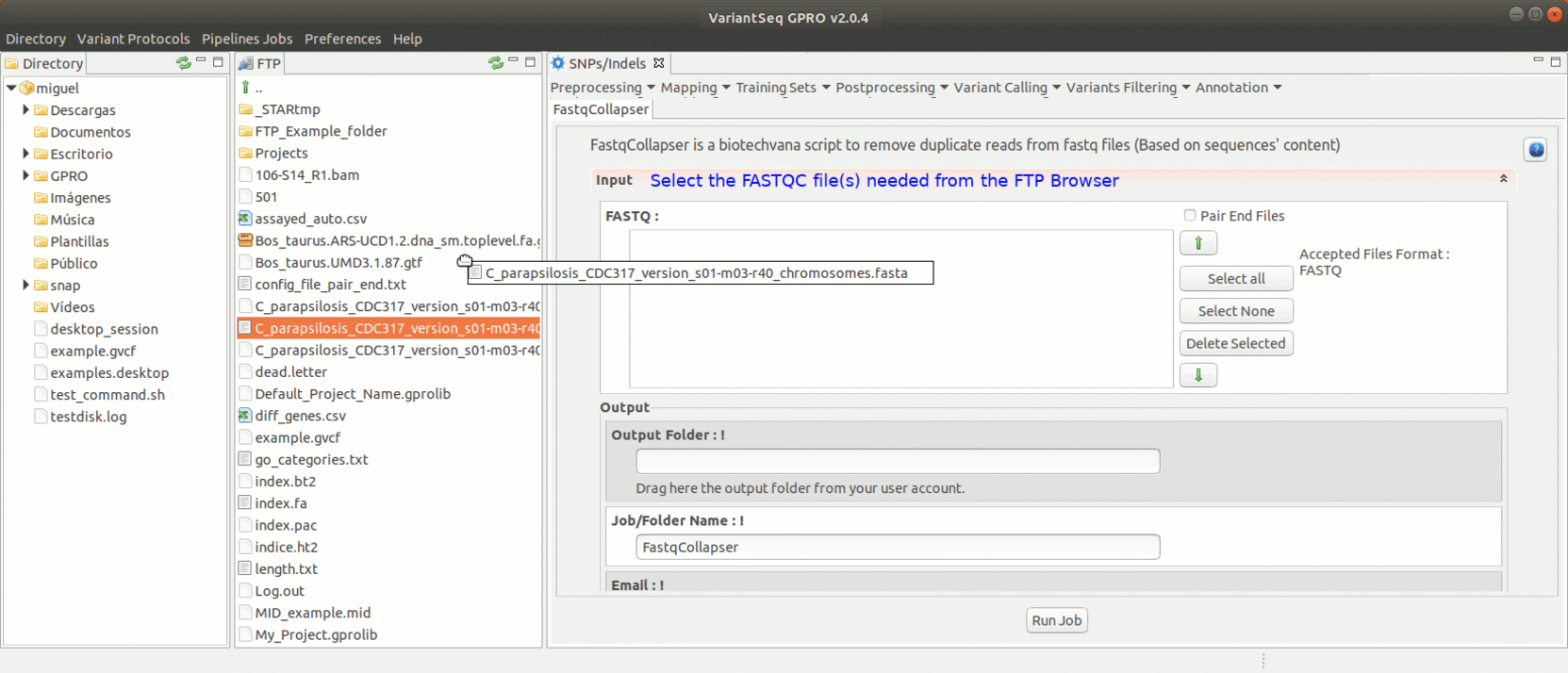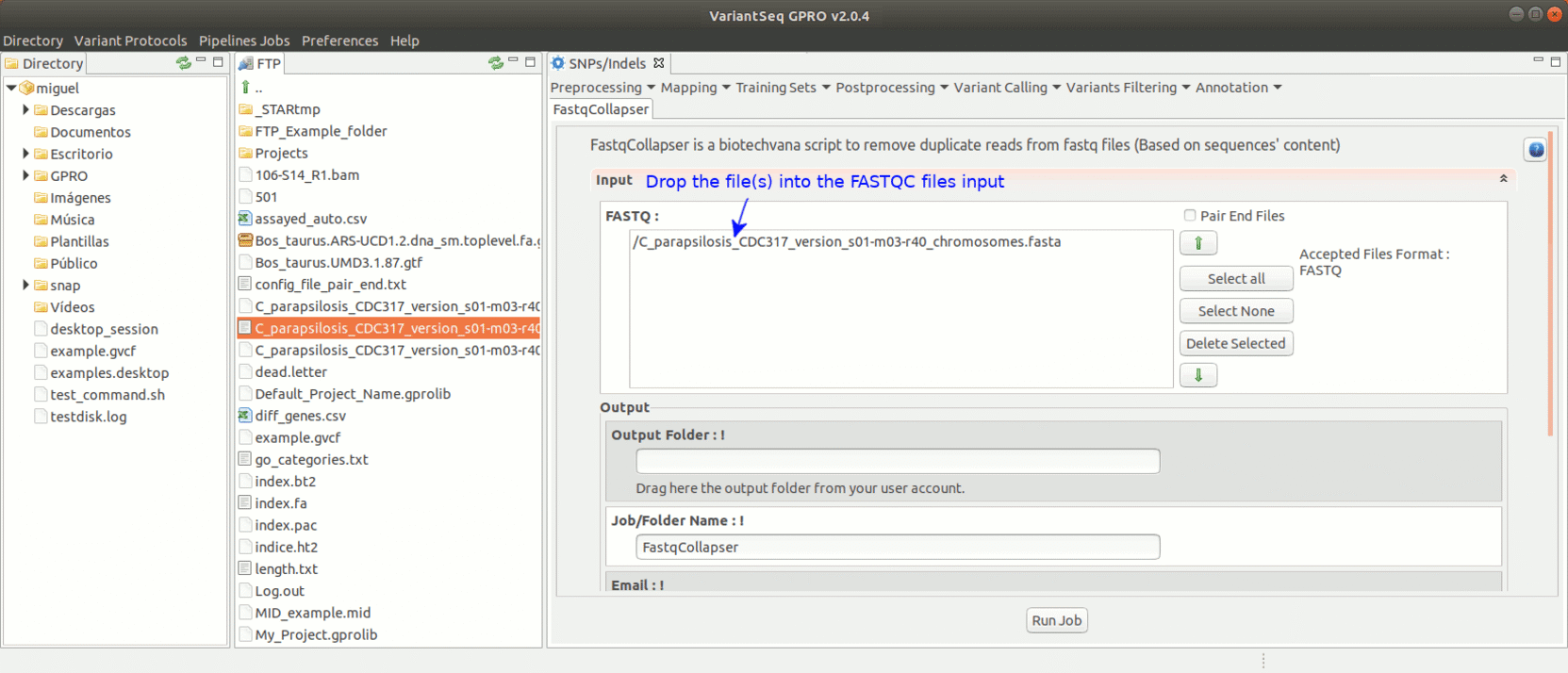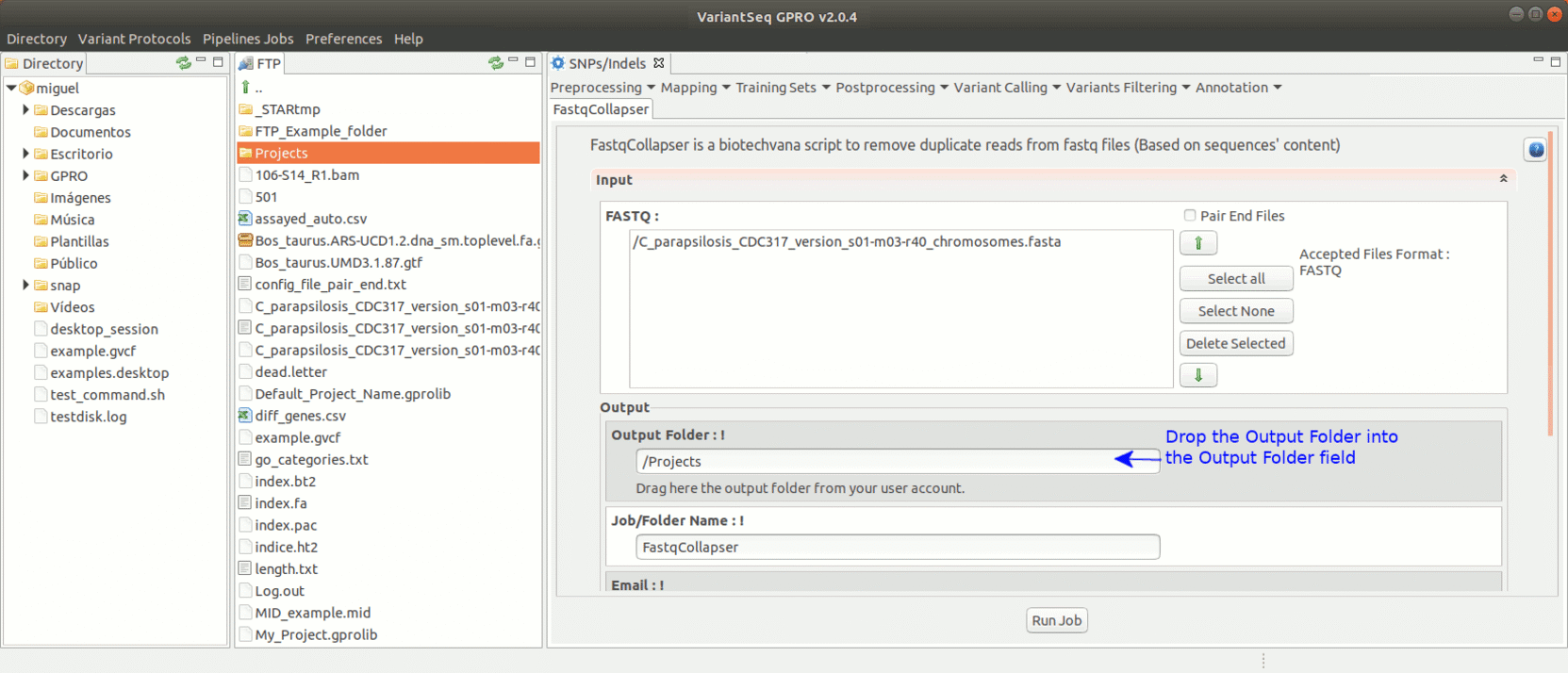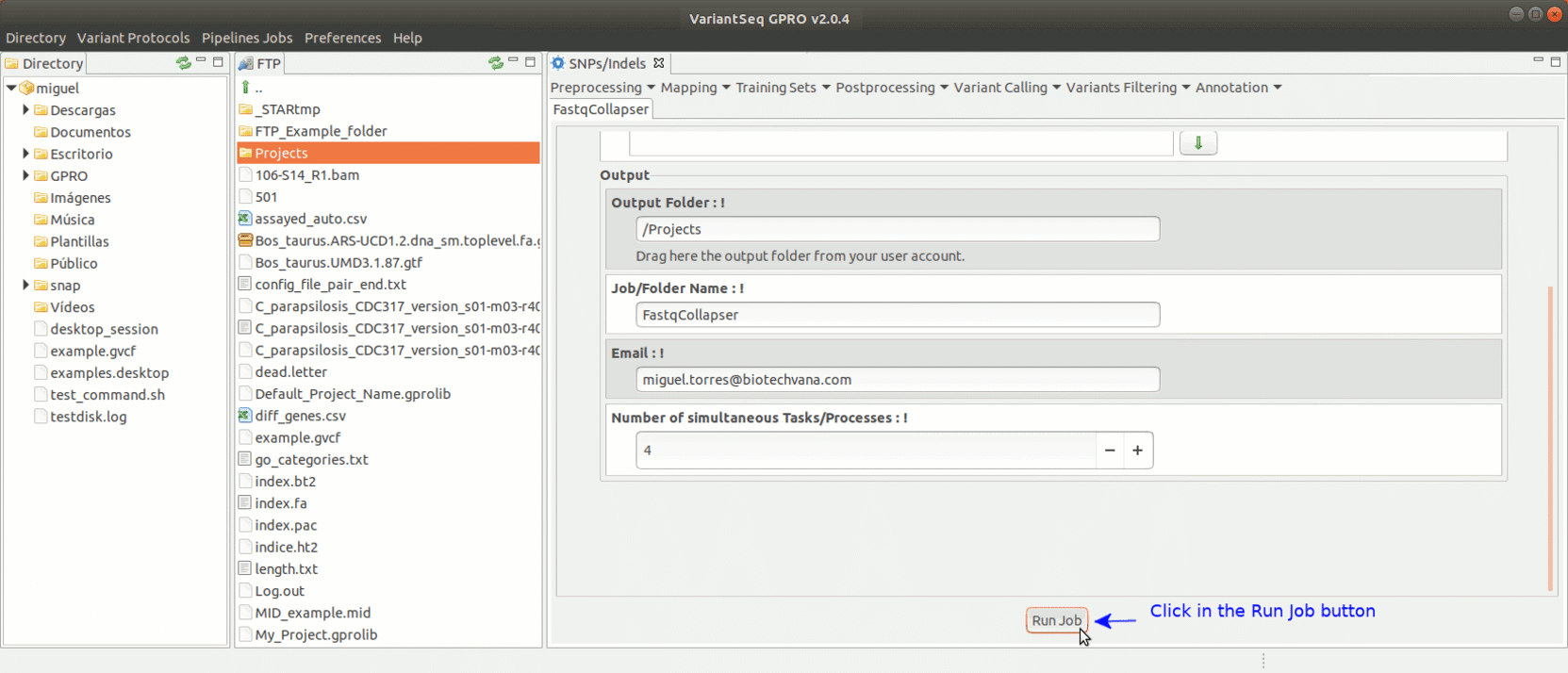
Figure 13: Using the GPRO interface for FastQCollapser.
- Trimming & Cleaning: FastqIntersect
FastqIntersect is a script that compares the information of two pair-end files that have been independently preprocessed and the information on both files to edit them keeping only those reads, and in the same order, that are present in both files (mate reads). This tool used when the number of reads obtained does not match the output of the execution of any preprocessing tool in each file individually the other. This is because assembly/mapping processes require that the files match in the number and the sort of reads. Please note that both Prinseq and Trimmomatic already have a function to intersect reads by ticking the ‘pair end files’ box. Thus, FastqIntersect will only need to be run in either those cases where the ’pair end files’ box has not been selected. FastqIntersect will also not need to be used when Cutadapt has been used, since this tool does not implement intersecting functions.
To run FastqIntersect, go to [ Preprocessing→ PrepSeq → FastqIntersect ] and proceed as shown in Fig. 14.
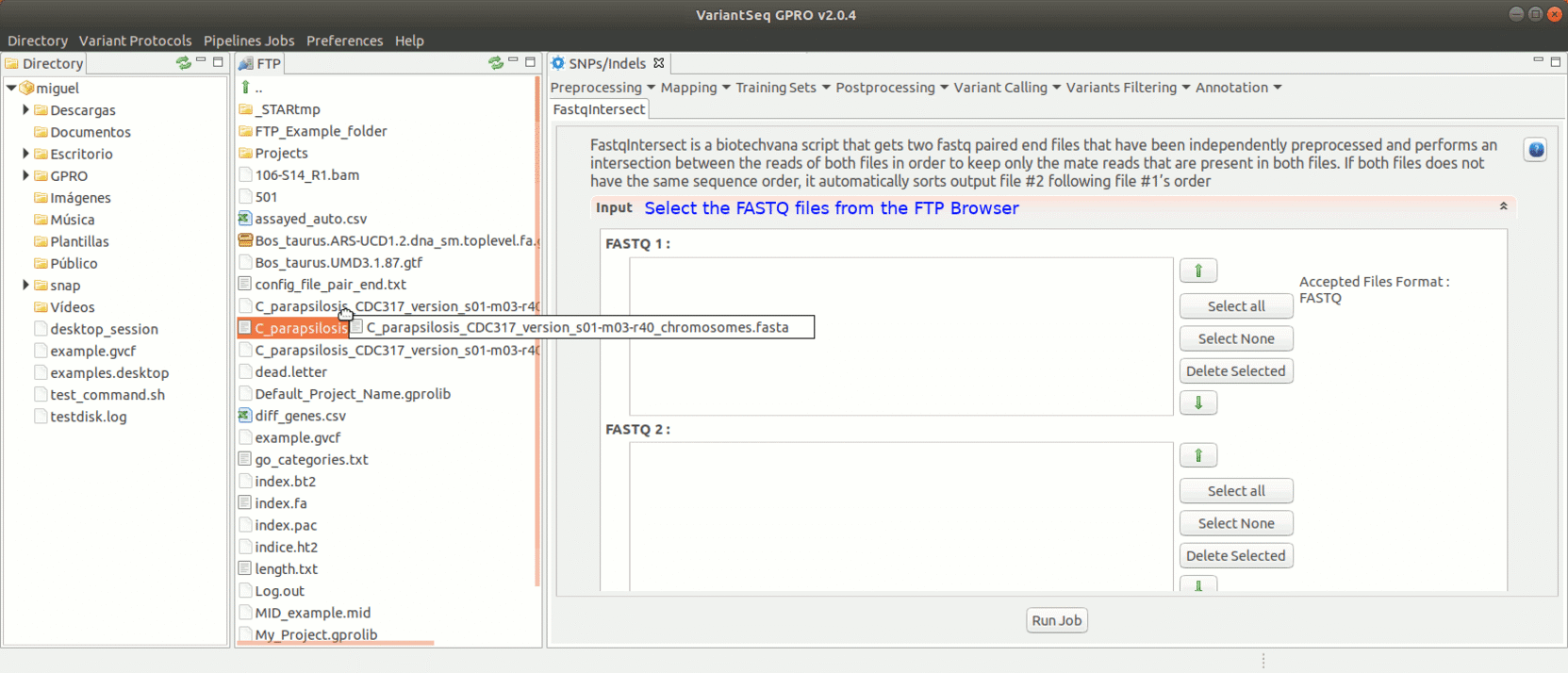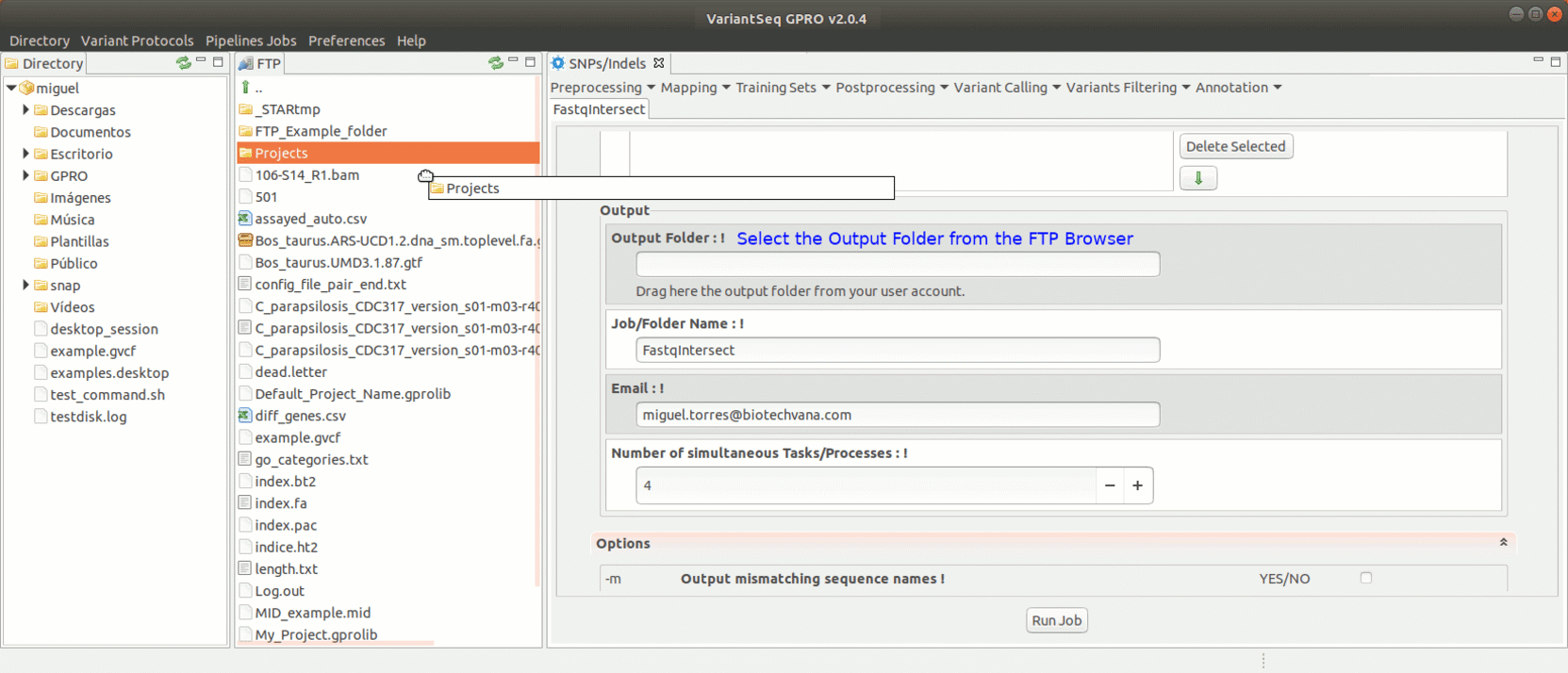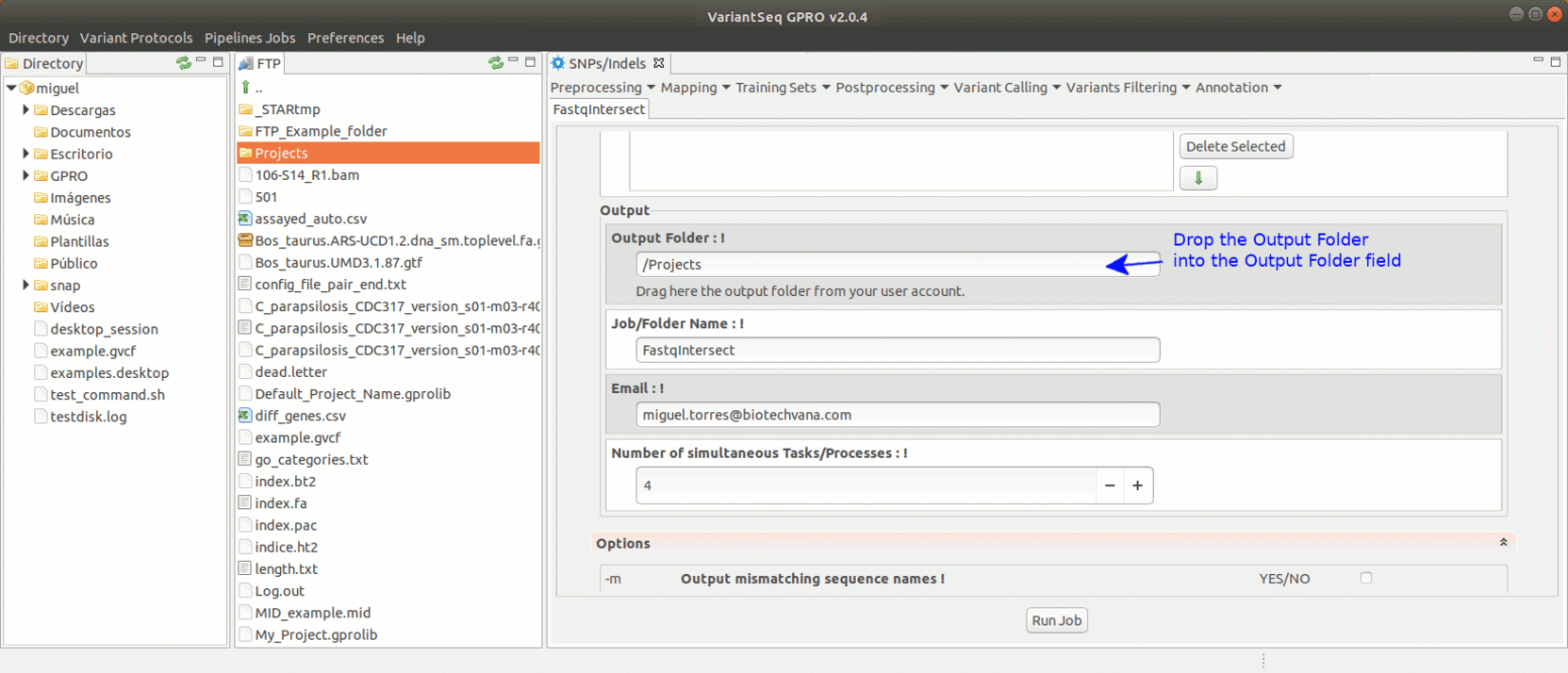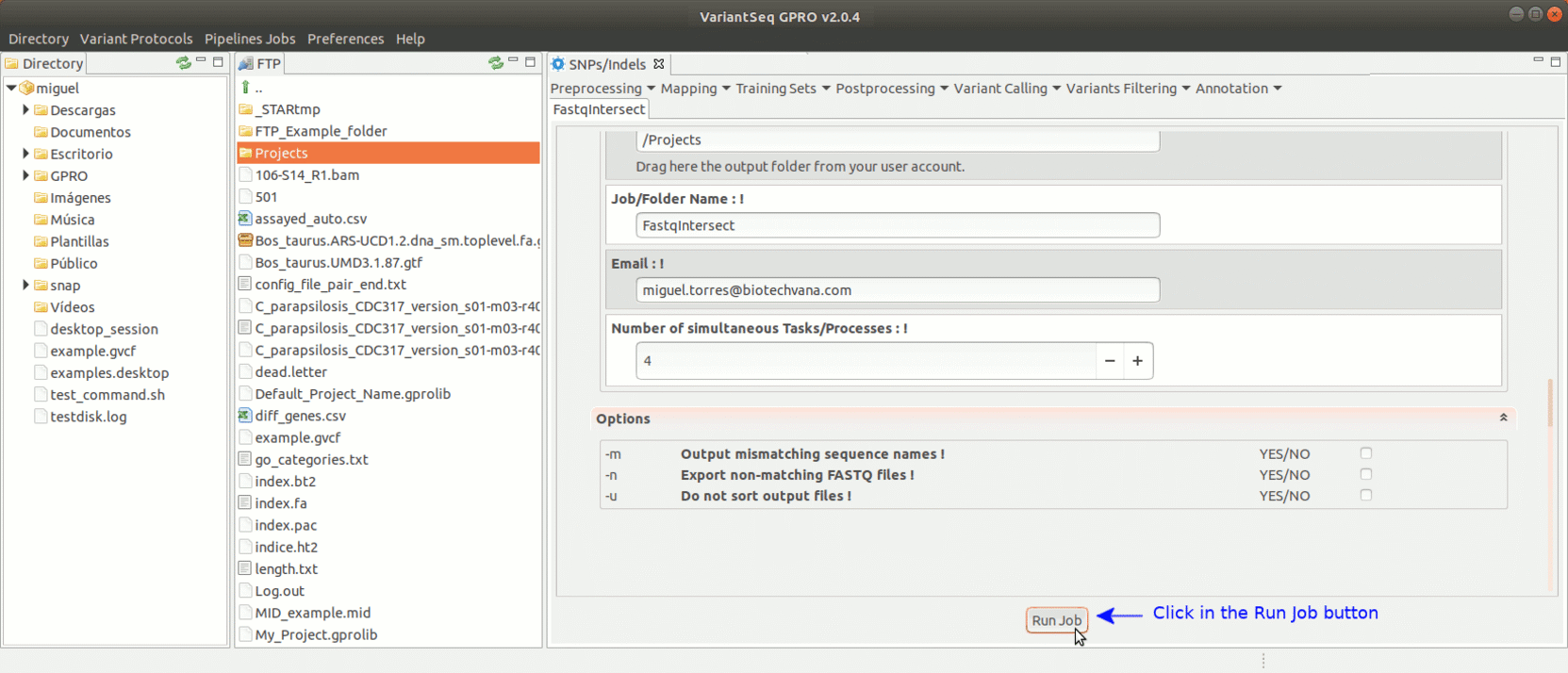
Figure 14: Using the GPRO interface for fastqintersect.
- Mapping: Bowtie2
Bowtie2 is an ultrafast and memory-efficient tool for aligning of sequencing reads onto long reference sequences. For further details on this tool see the Bowtie2 manual at http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml.
To run Bowtie2 go to [ Mapping→ Bowtie2 ] and follow Fig. 15.
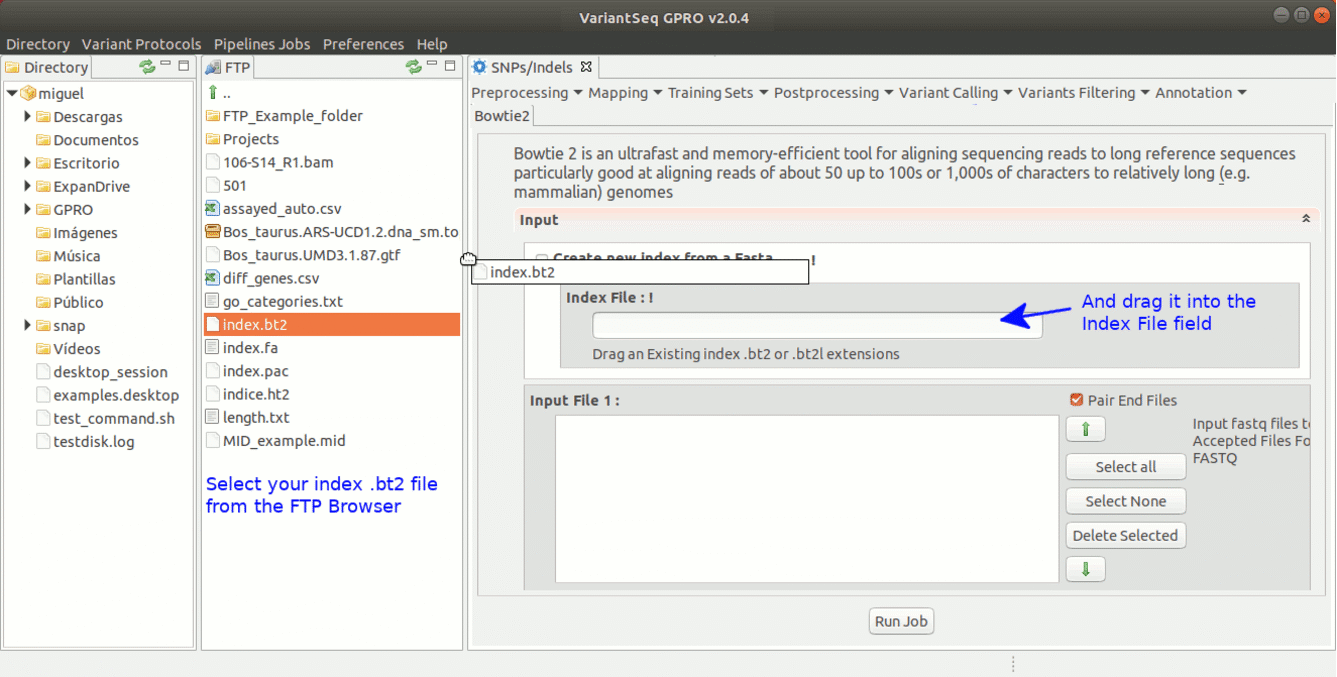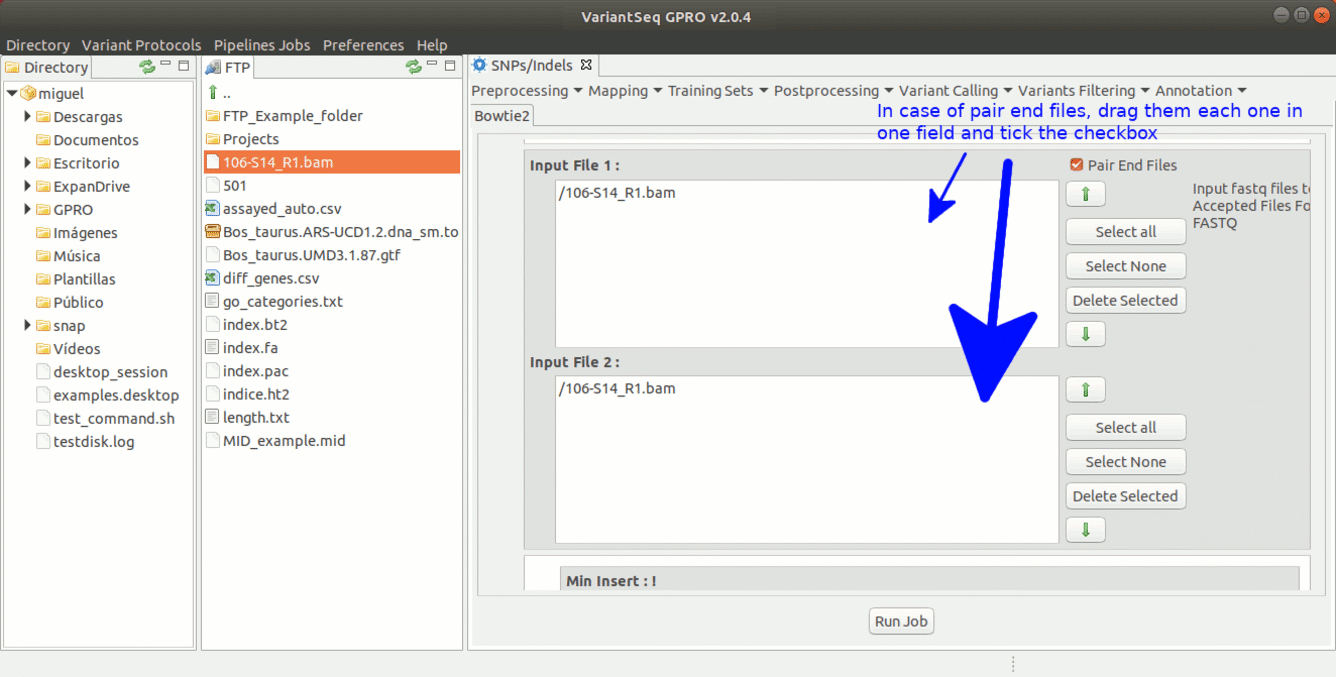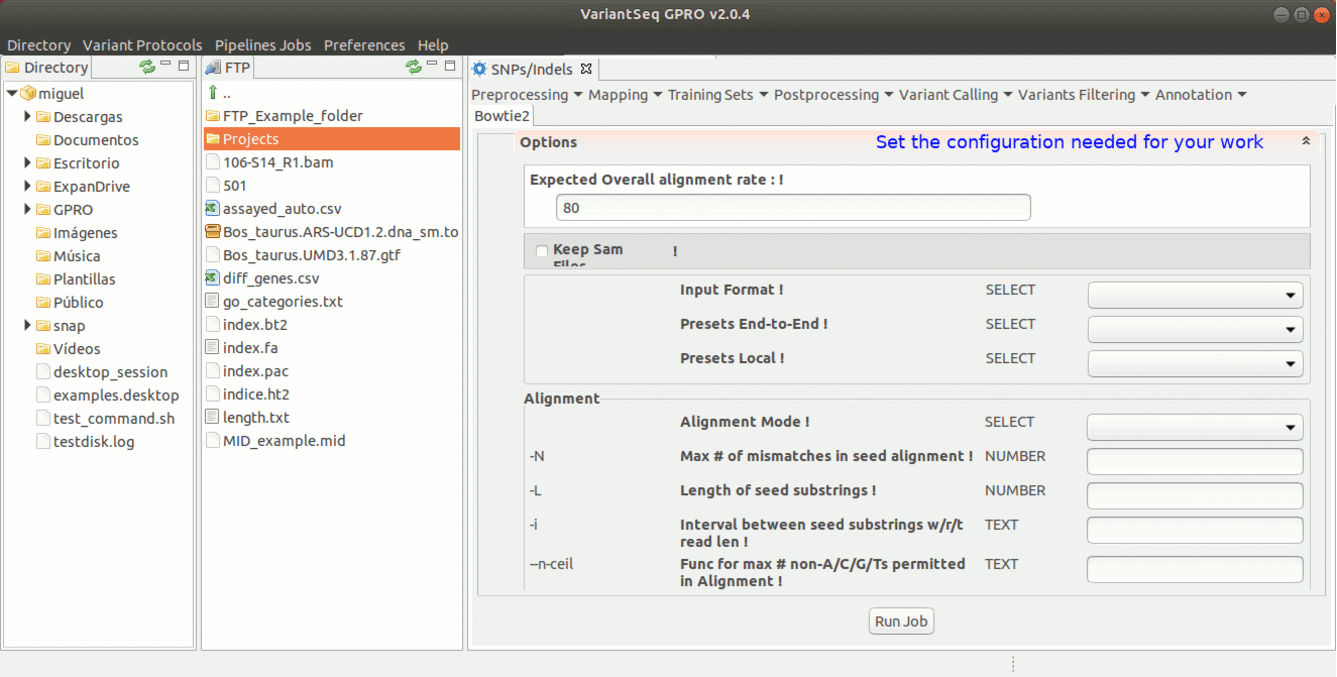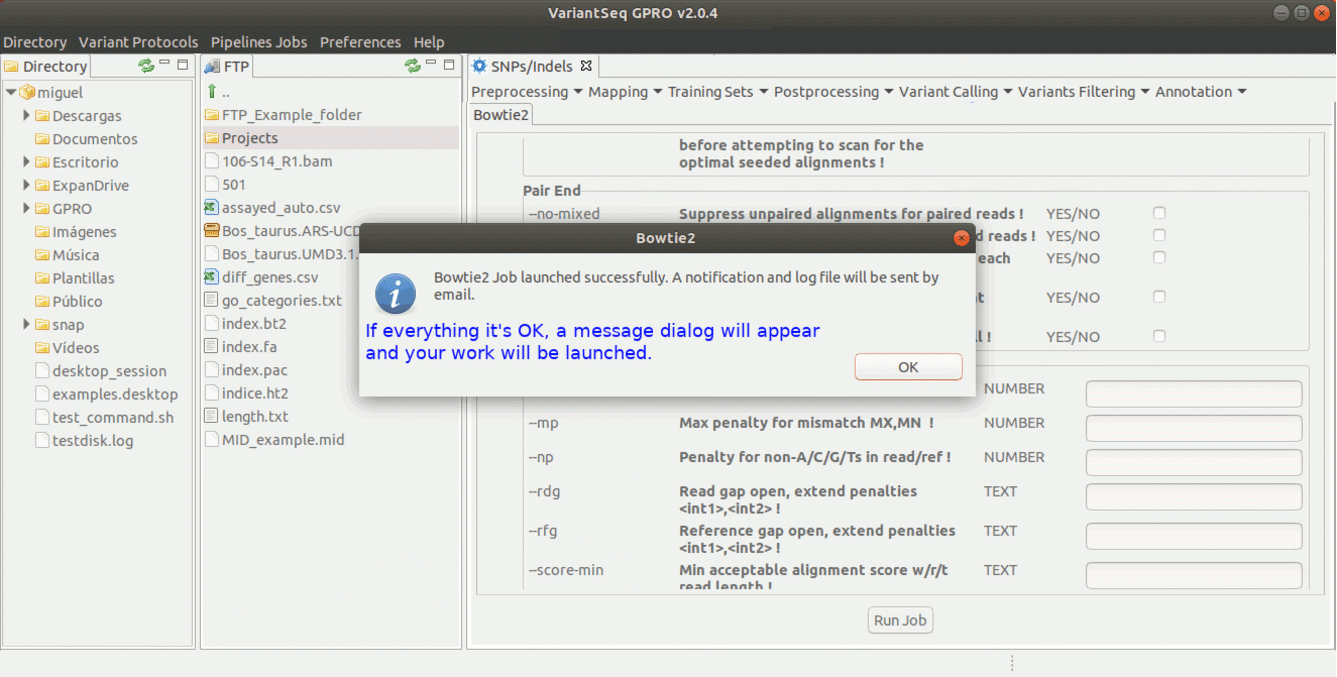
Figure 15: Using the GPRO interface for Bowtie2.
- Mapping: BWA
BWA is a software package for mapping of low-divergent sequences against large reference genomes. It consists of three different algorithms, namely: BWA-backtrack, BWA-SW, and BWA-MEM. The first algorithm is specifically designed for Illumina sequence reads up to 100bp in length, while the other two are implemented for sequences ranging from 70bp to 1Mbp. BWA-MEM and BWA-SW share features such as long-read support and split alignment. BWA-MEM, however, is generally recommended for high-quality queries as it is faster and more accurate. For more information, see the BWA manual at http://bio-bwa.sourceforge.net/bwa.shtml.
To run BWA go to [ Mapping → Bwa ] and follow Fig. 16.
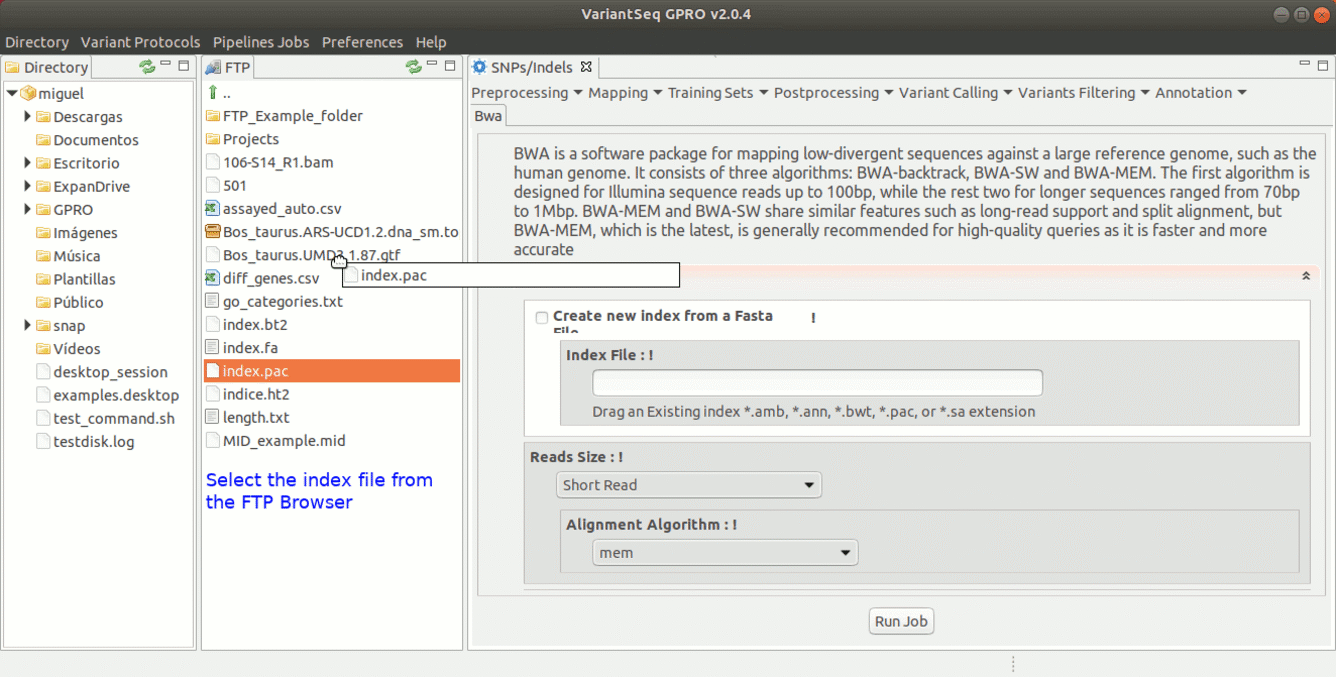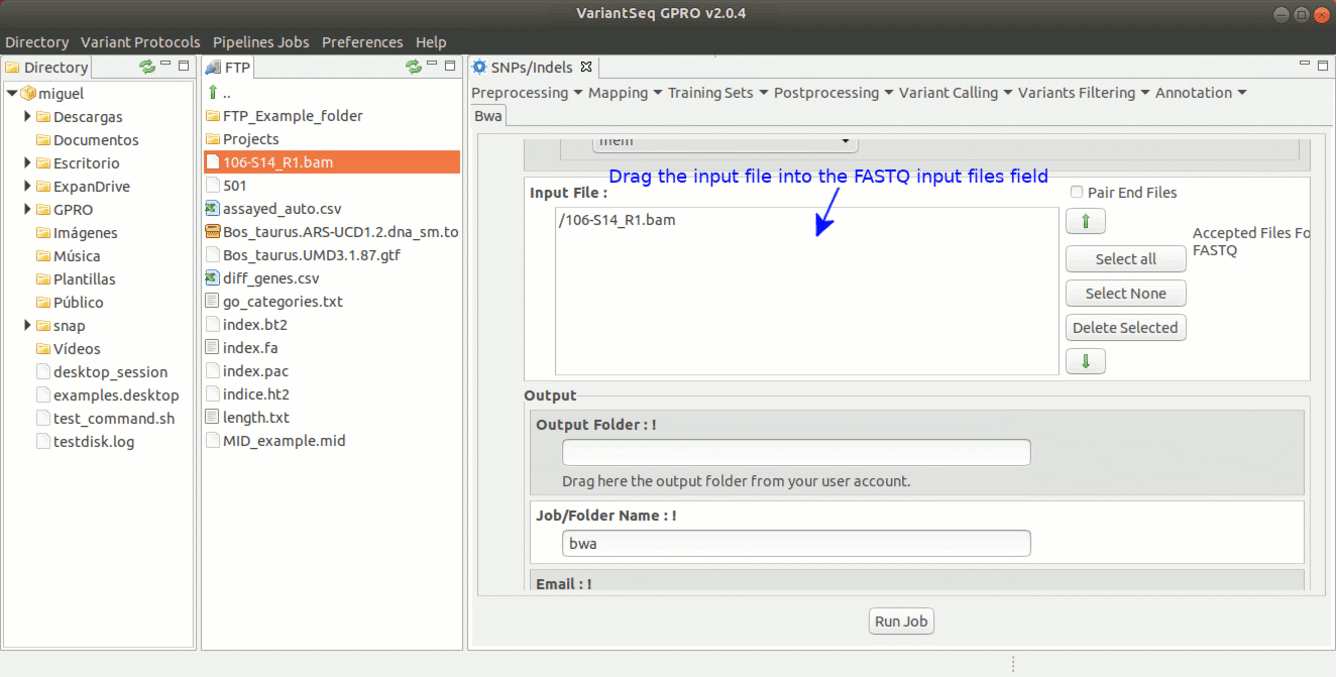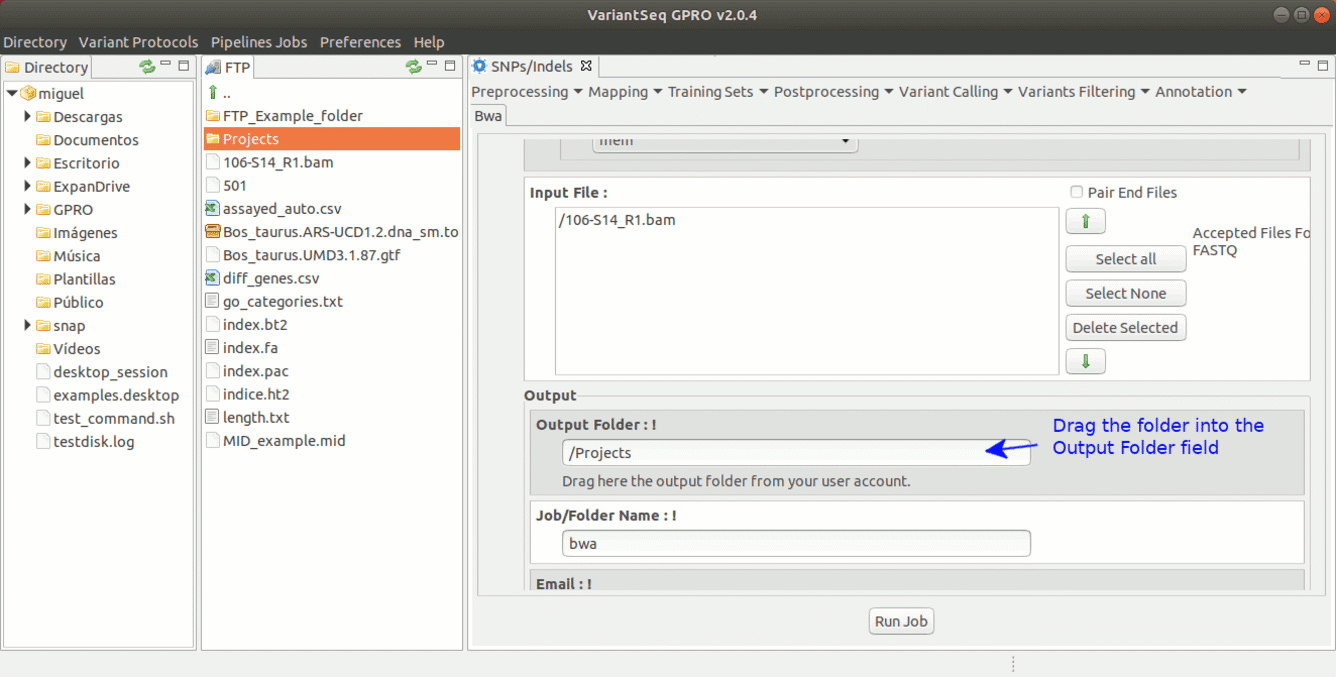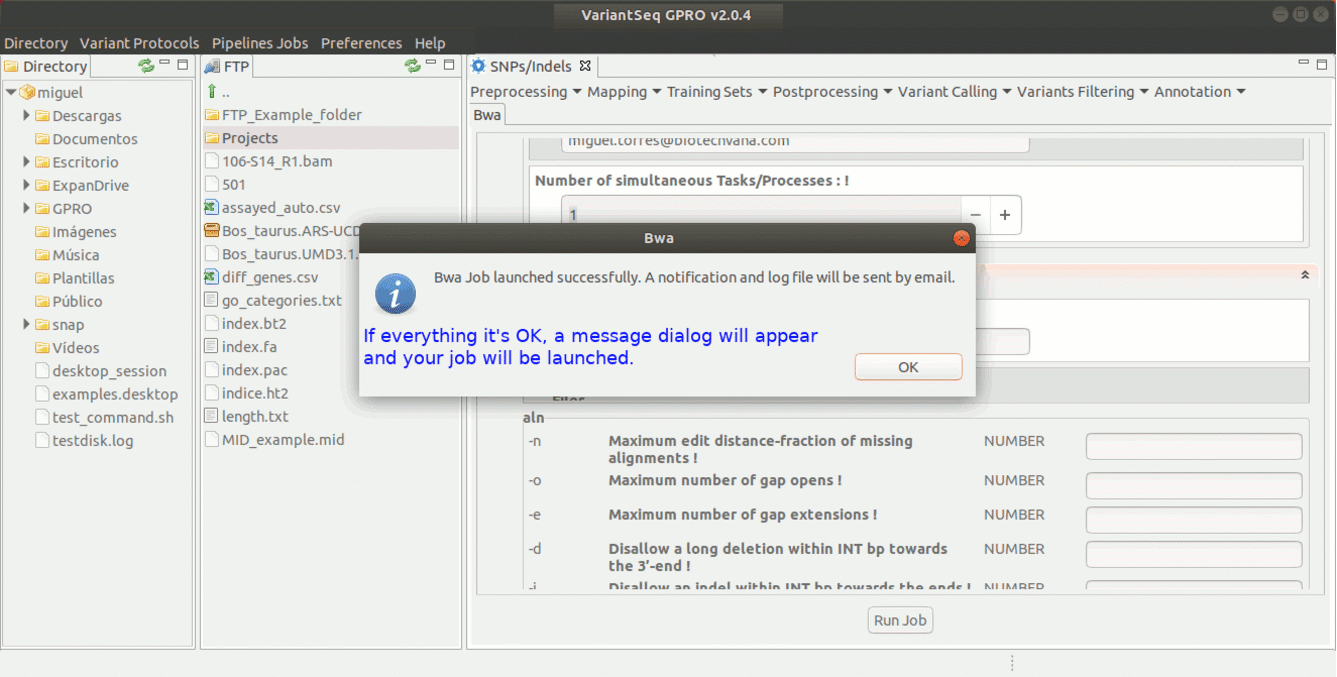
Figure 16: Using the GPRO interface for BWA.
- Mapping: Hisat2
HISAT2 (Kim, Langmead, and Salzberg 2015) is tool for mapping of sequencing reads of both DNA and RNA. For more information, see the HISAT2 manual at Hisat2 manual.
To run Hisat2, go to [ Mapping → Hisat2] and follow Fig. 17.
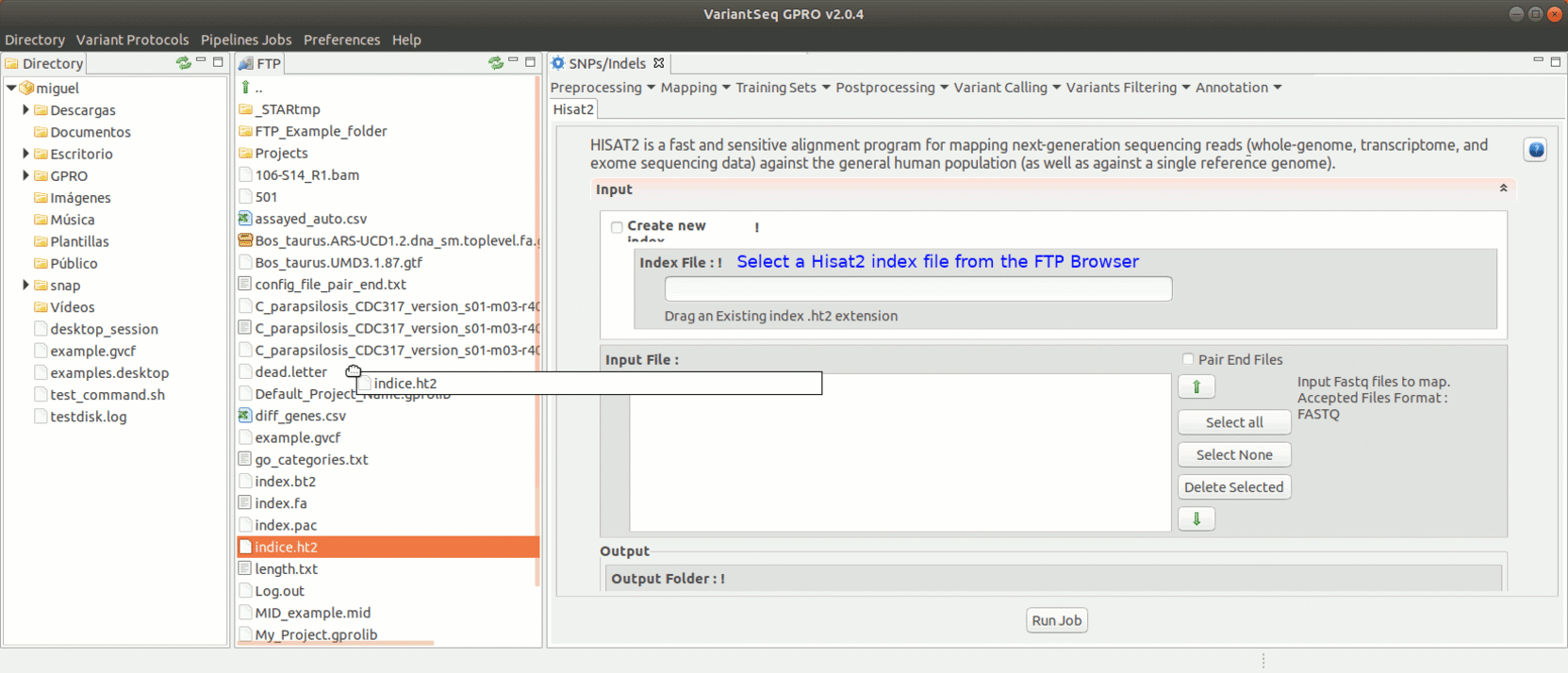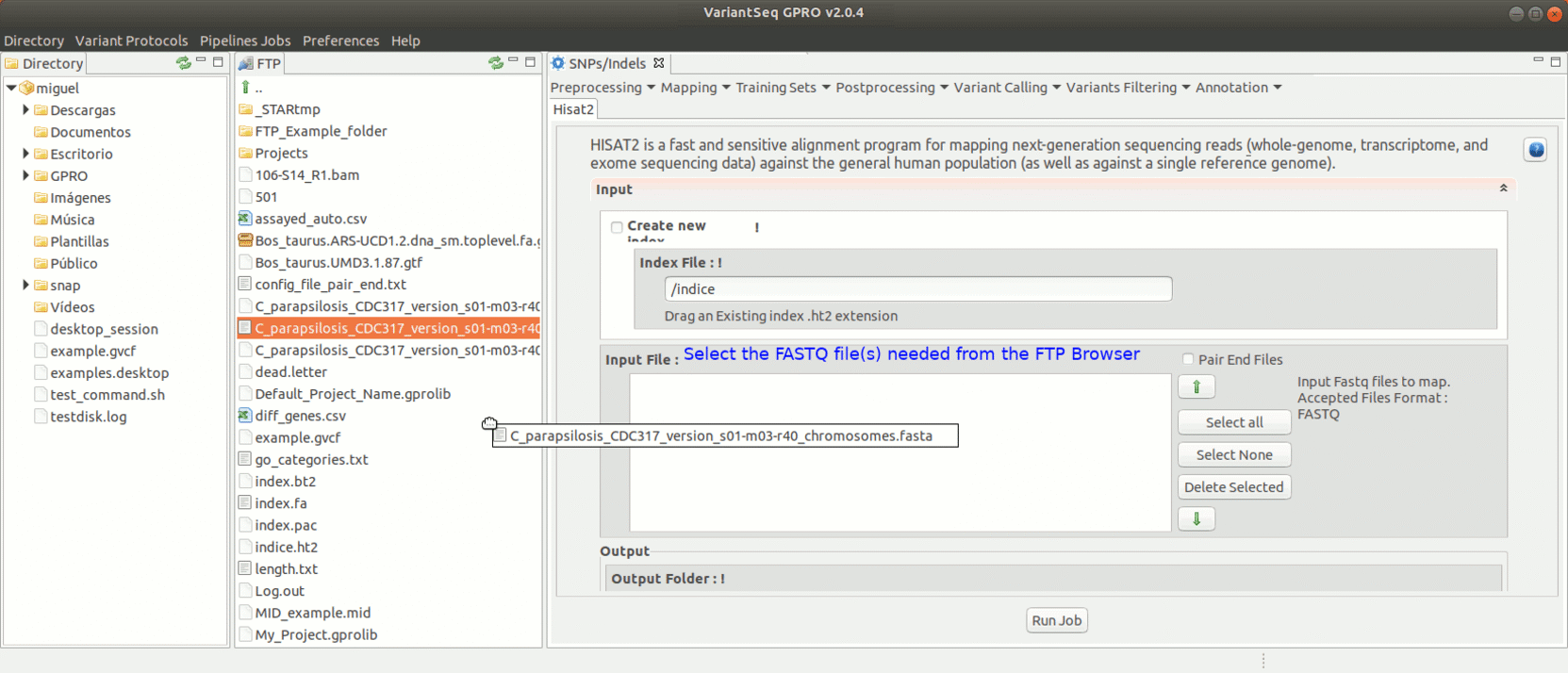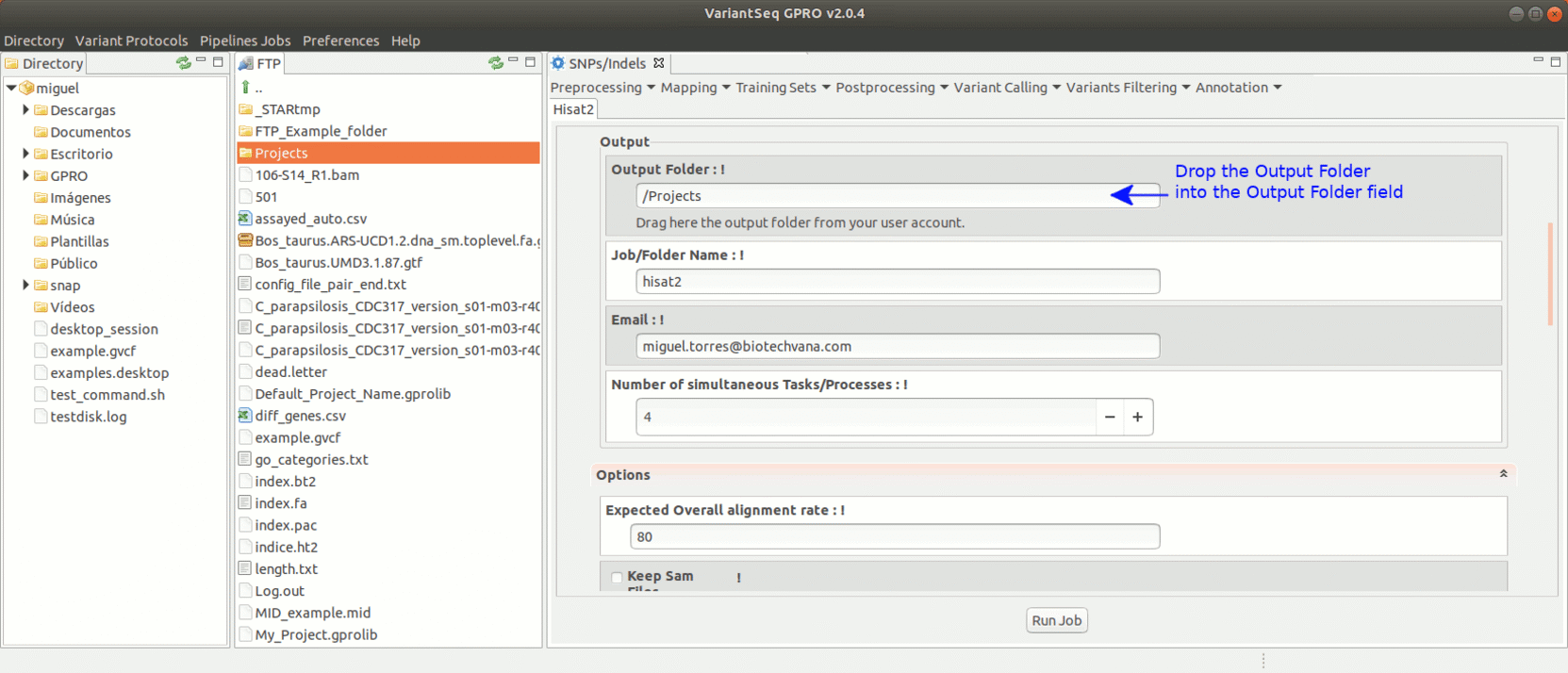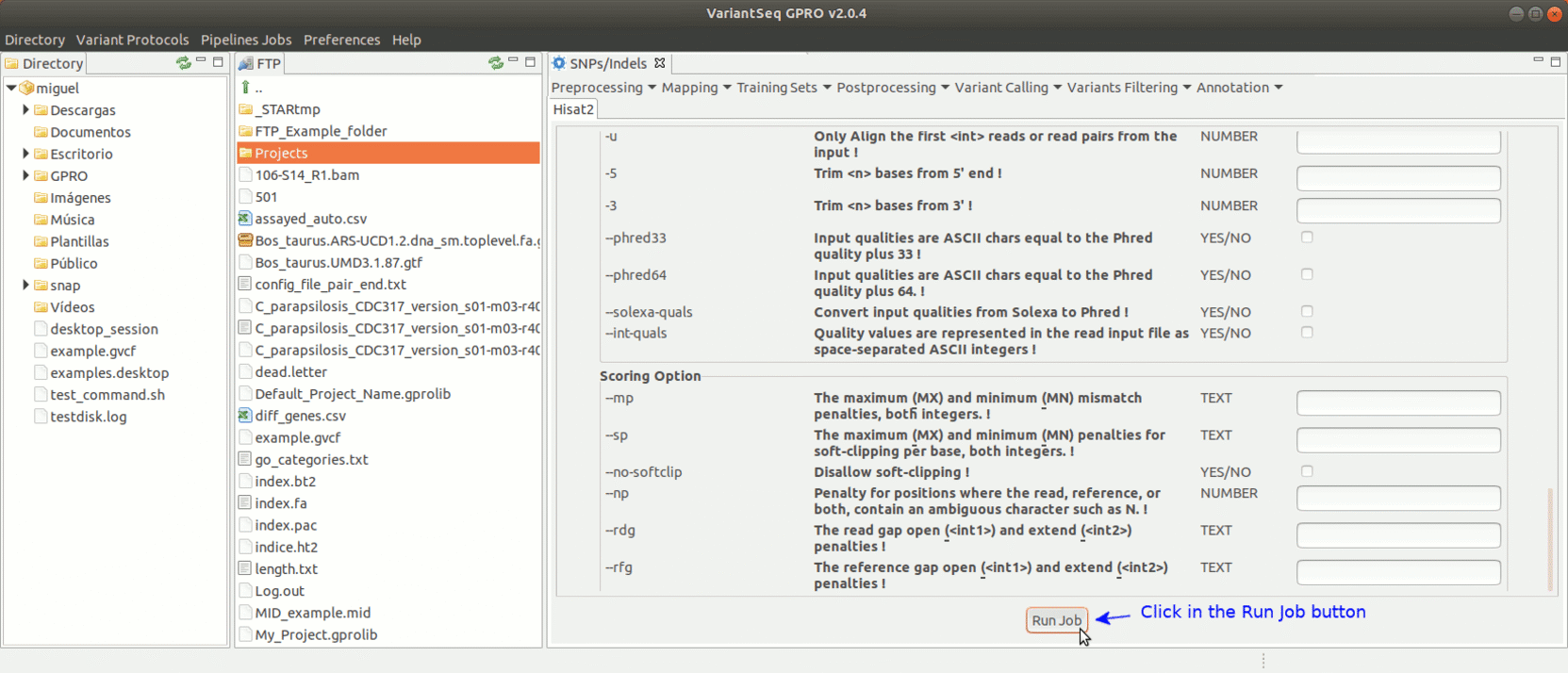
Figure 17: Using the GPRO interface for Hisat2.
- Mapping: Tophat
TopHat aligns RNA-Seq reads to a reference genome identifying exon-exon splice junctions. For more information, see the Tophat manual at Tophat manual .
To run Tophat, go to [Mapping → Tophat ] and follow Fig. 18.
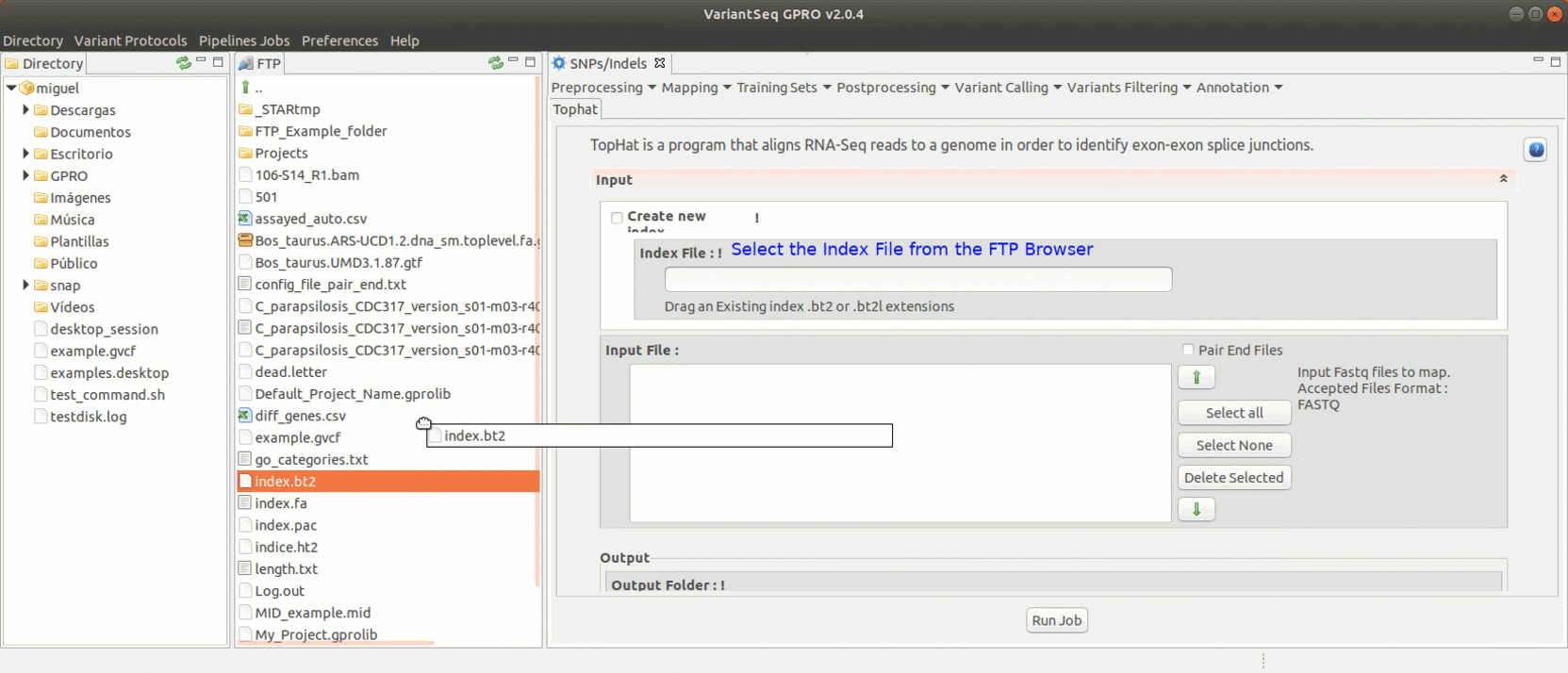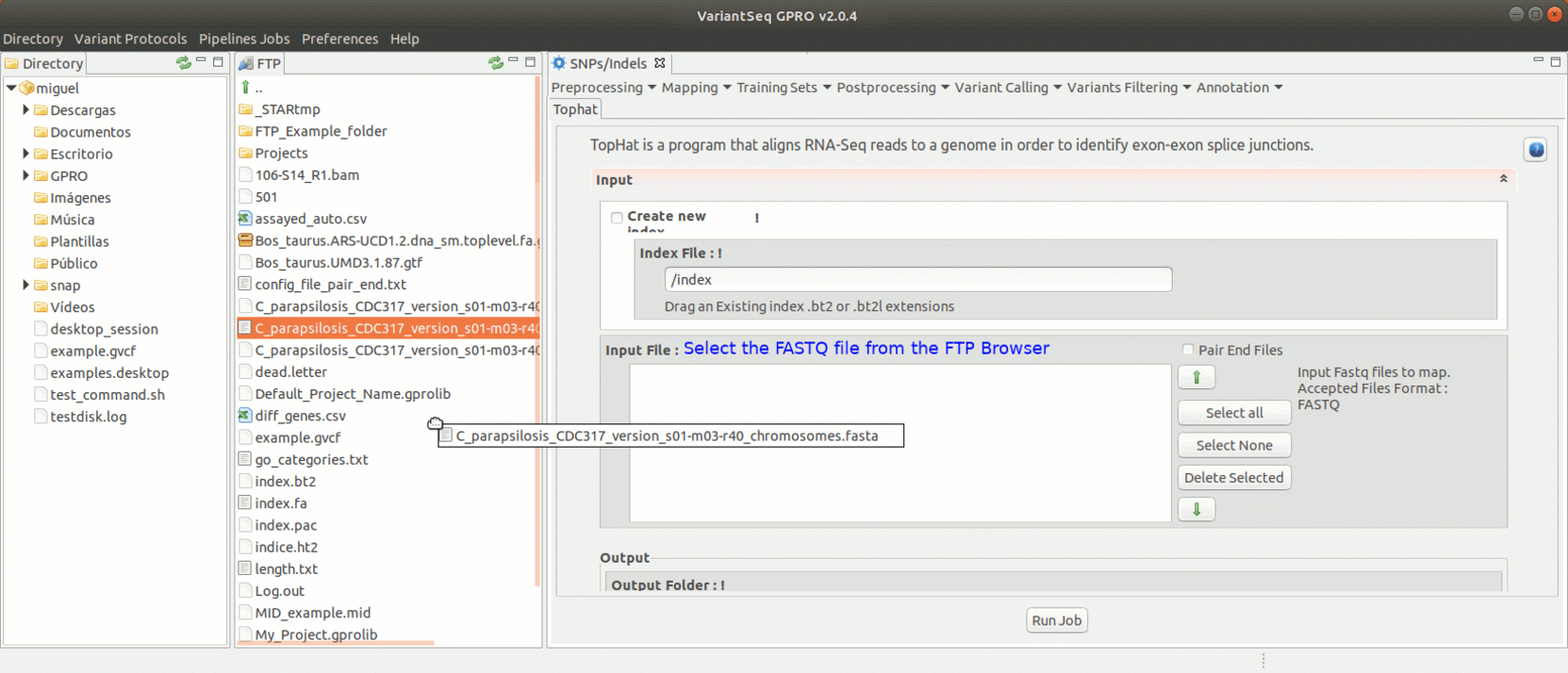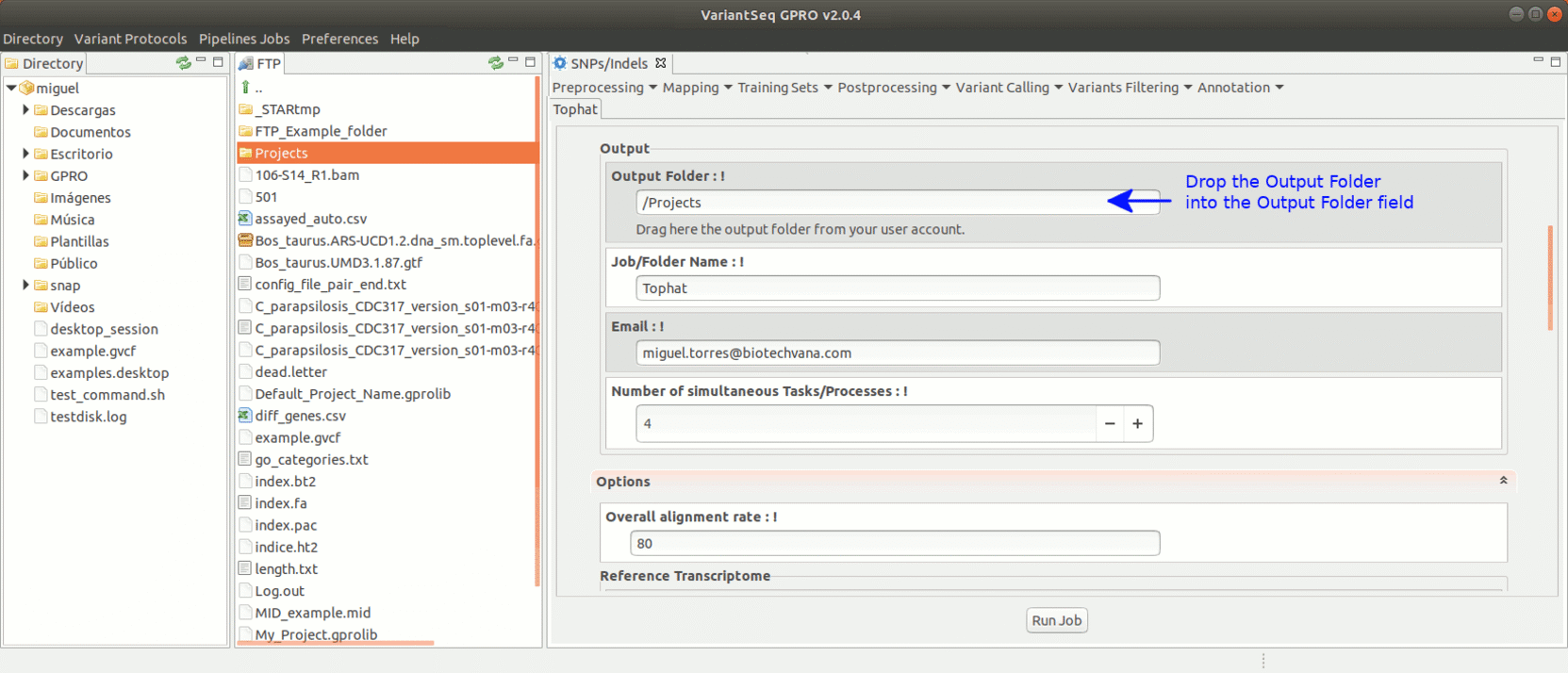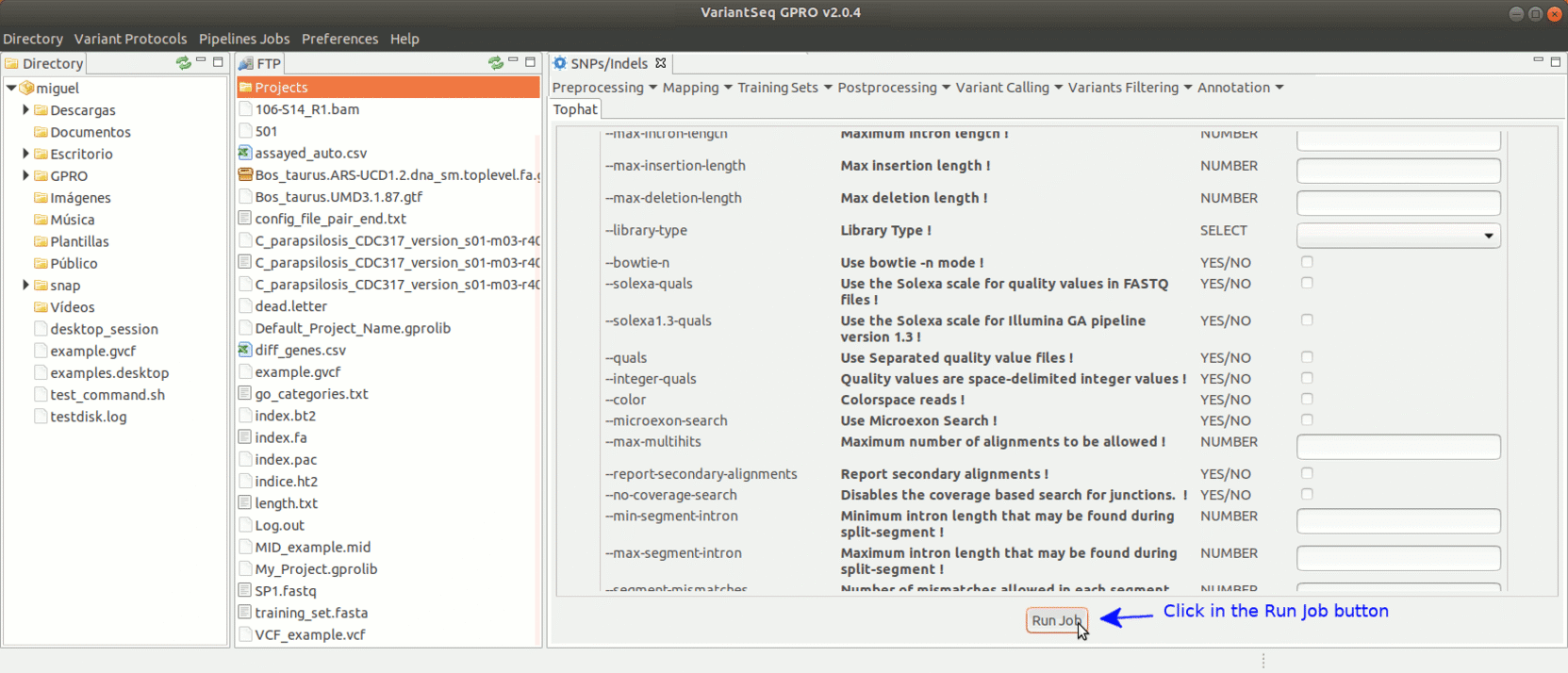
Figure 18: Using the GPRO interface for TopHat.
- Mapping: STAR
STAR (Dobin et al. 2013) is a universal aligner for mapping RNAseq spliced reads and transcripts. For more information, see https://github.com/alexdobin/STAR.
To run STARS, go to [Mapping → STARS ] and follow Fig. 19.
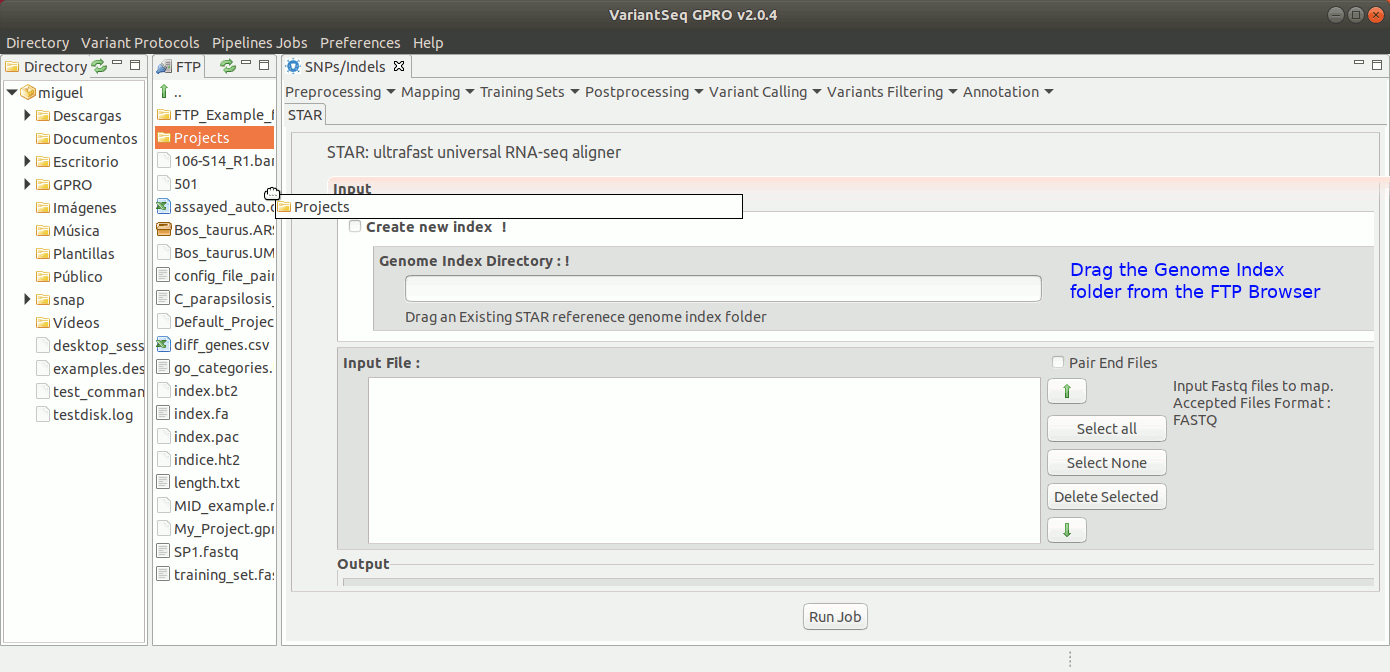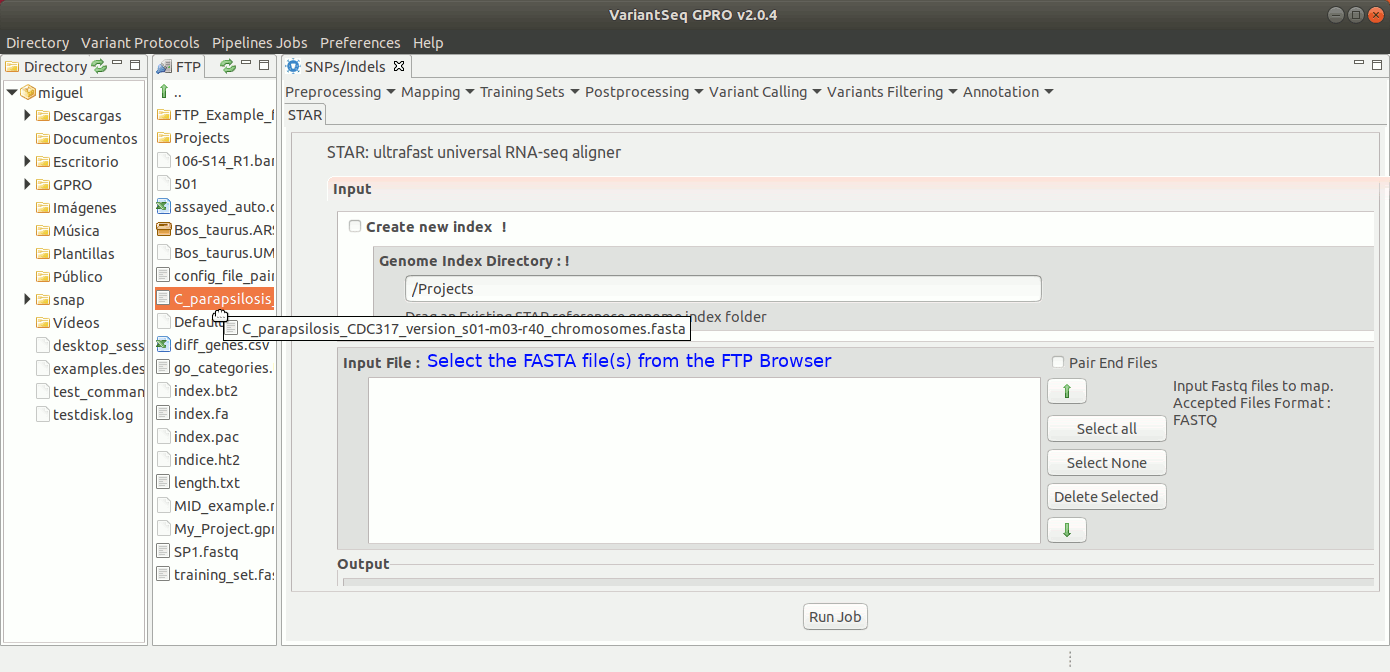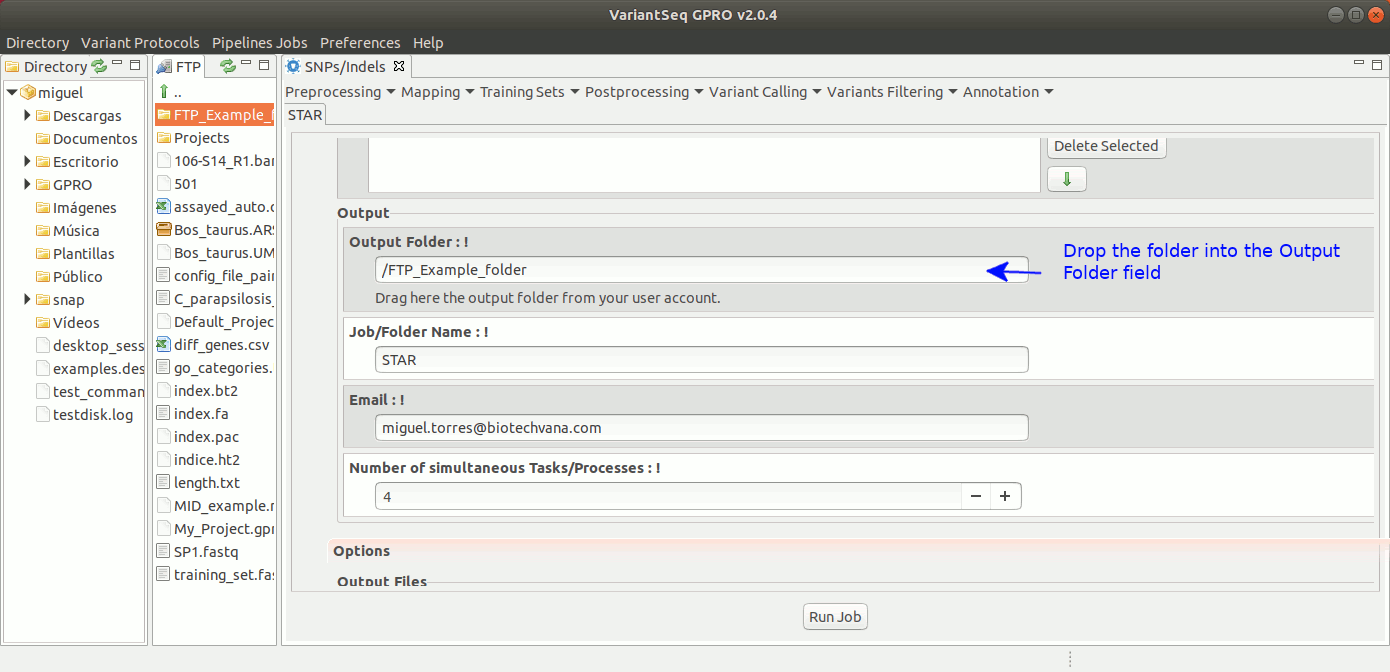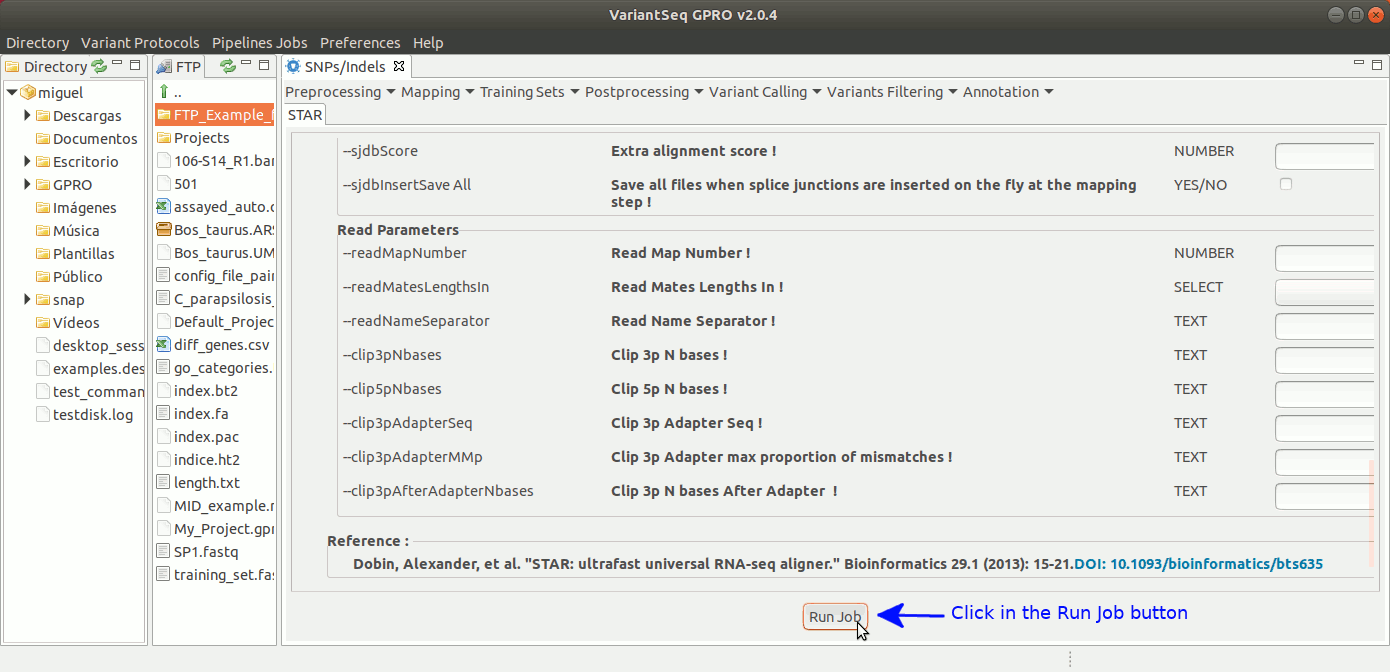
Figure 19: Using the GPRO interface for STARS.
- Training Resources: Generate Training Sets
Many GATK tools need sets of known variants resources (Known Variants, Training sets, and/or Truth Sets) to distinguish true variants from false positives and artifacts. See the GATK forum at https://software.broadinstitute.org/gatk/documentation/article?id=11065 for more details. For genomes characterized from model organisms, training resources can be found in the repositories: GATK bundle resources, NCBI, Ensembl etc… However, if your project does not have a reference genome and associated training resources, VariantSeq provides a script running distinct Picard ( Wysoker et al. 2011 ), GATK (McKenna, 2010, DePristo, 2011 ) and SAM Tools (Li et al. 2009) to generate your own training set using your reference genome and the bam files generated after mapping your reads against that reference.
To generate a Training Set, go to [SNP/Indels → Training Resources → Generate Training Set] and follow Fig 20.
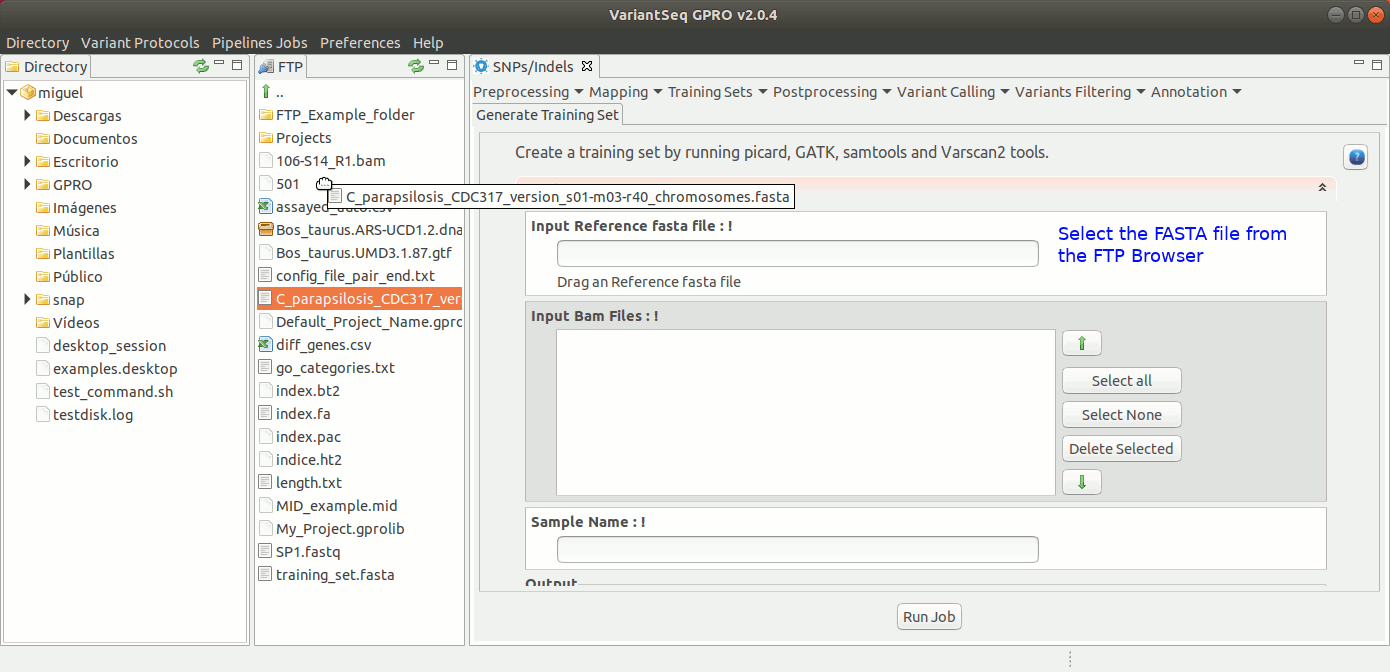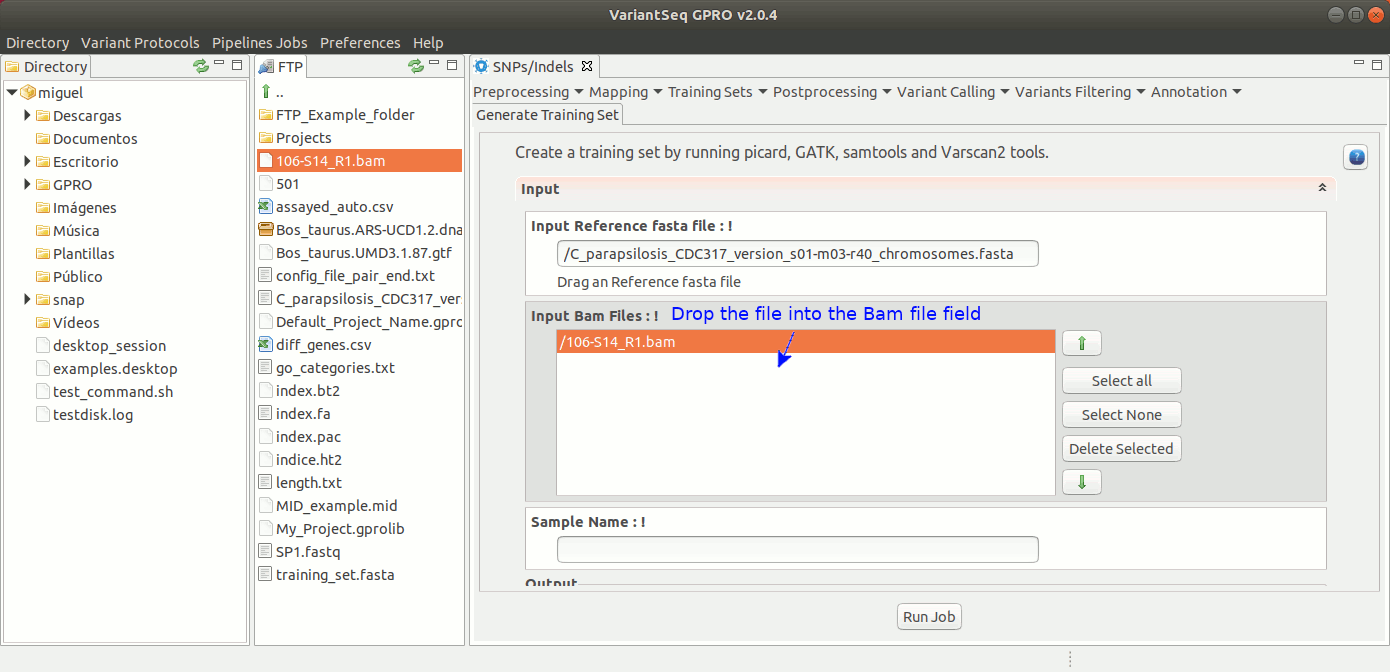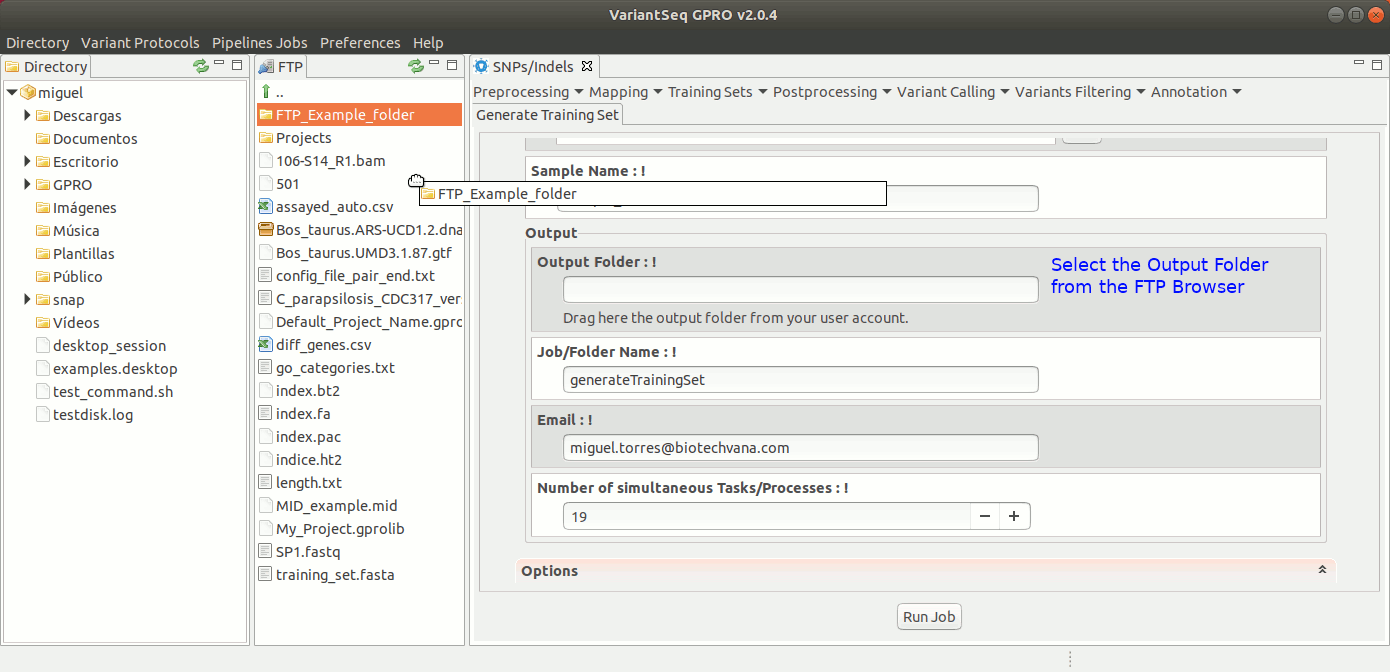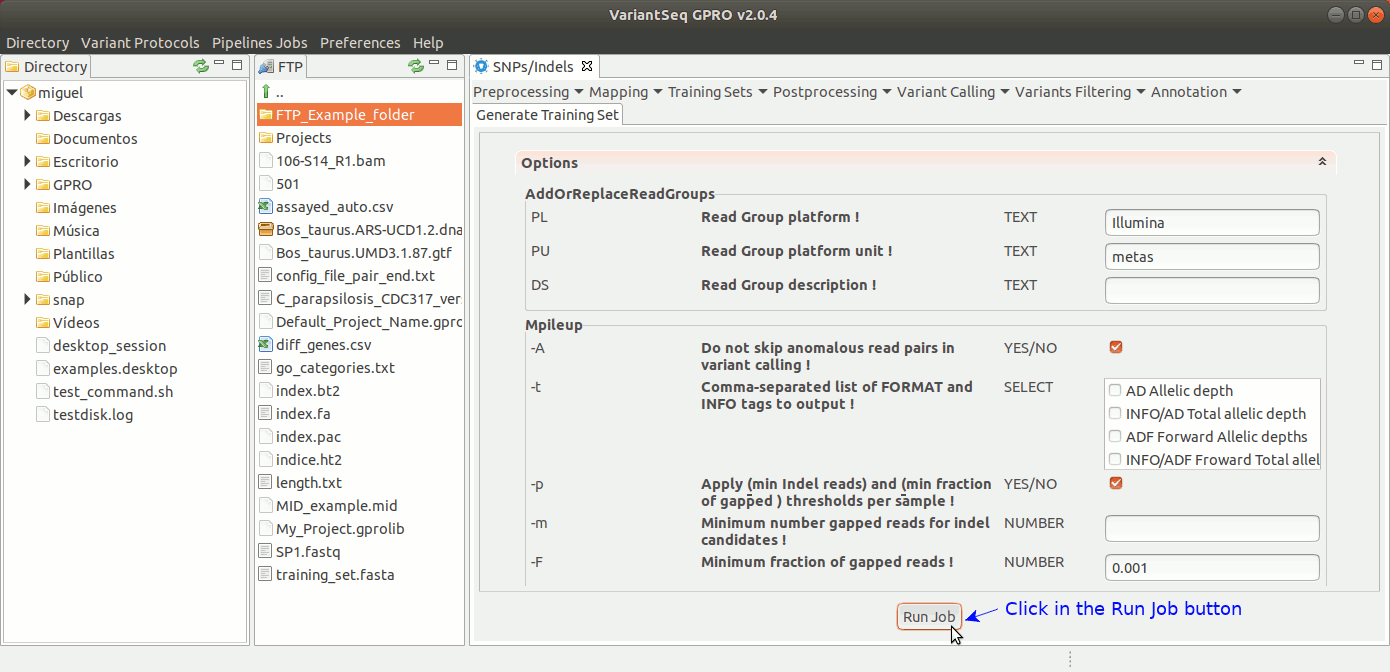
Figure 20: Using the GPRO interface for using the Training set application.
- Training resources: Create Somatic Panel of Normals
Somatic variants related to cancer usually present extremely low allele-frequencies due to contamination of normal cells or because of tumor heterogeneity. VariantSeq allows you to create a panel of normals (PON) obtained from multiple normal sample callsets using Mutec2 of GATK (McKenna, 2010, DePristo, 2011) to filter germline and artifactual sites. For more details see this thread on the GATK Forum:To use Mutect2 to generate a PON, go to [ SNP/Indels → Training Resources → GATK Create Somatic Panel of Normals ] and follow Fig. 21
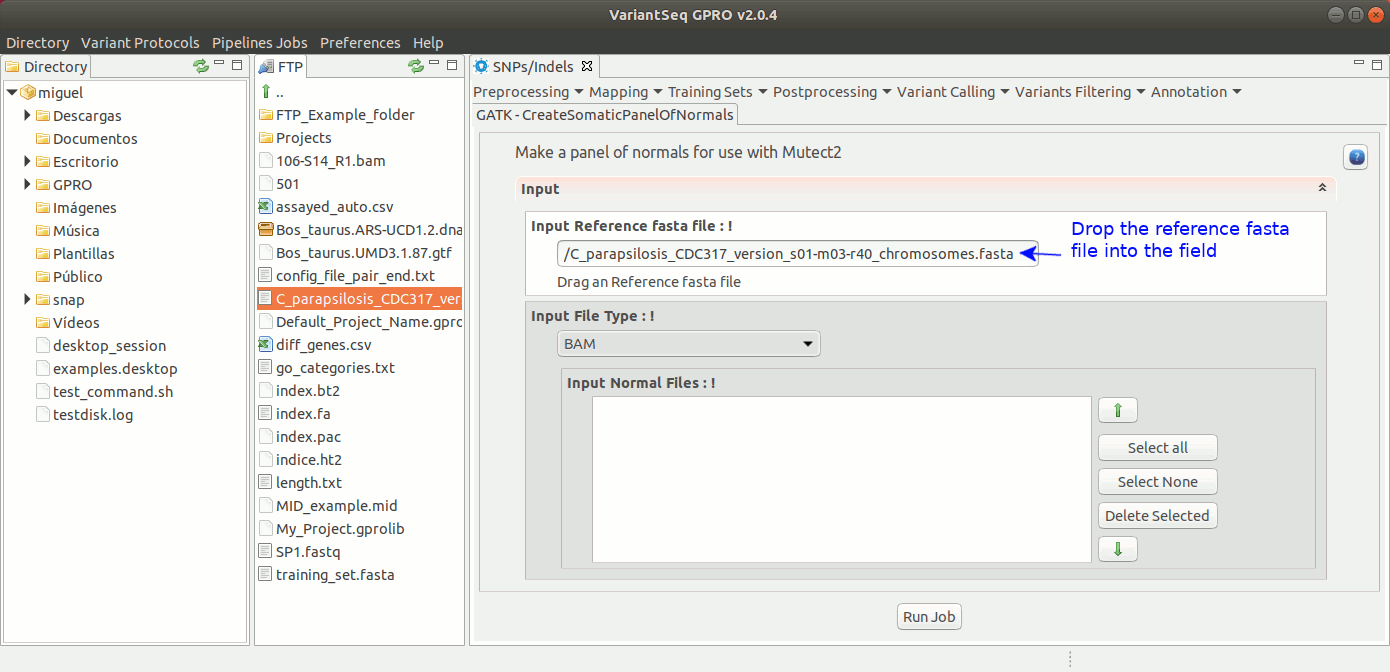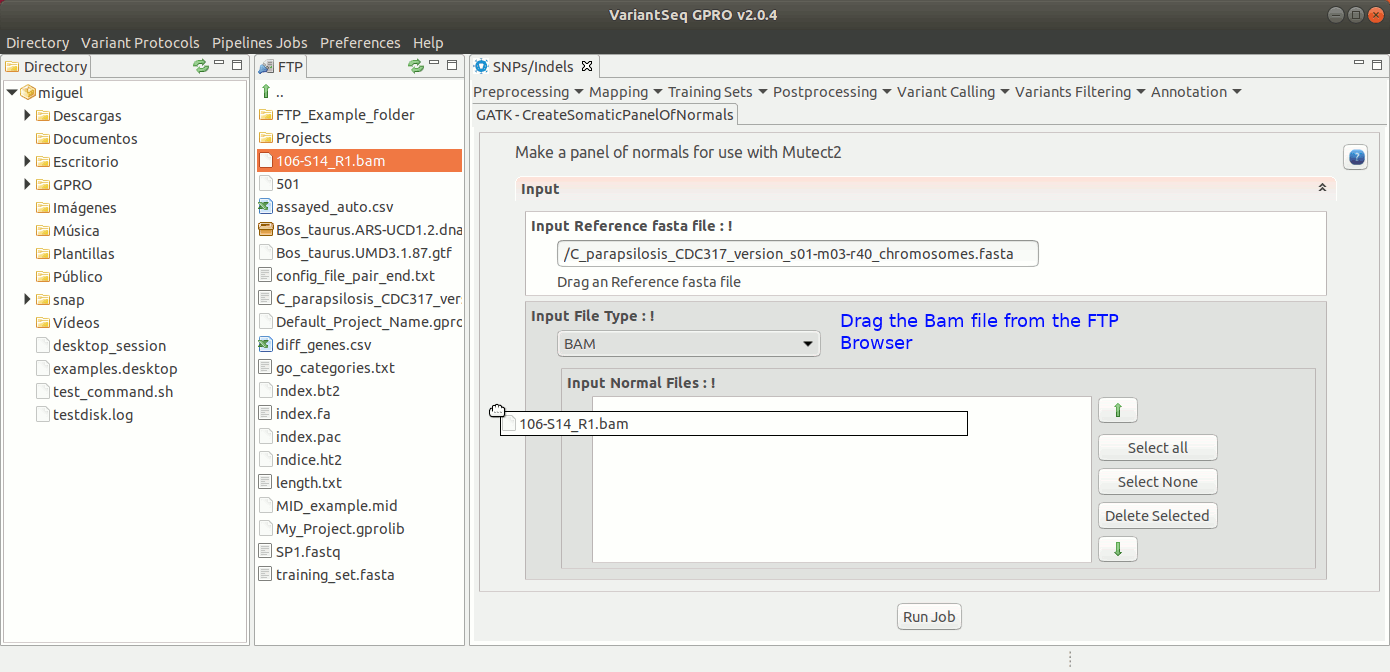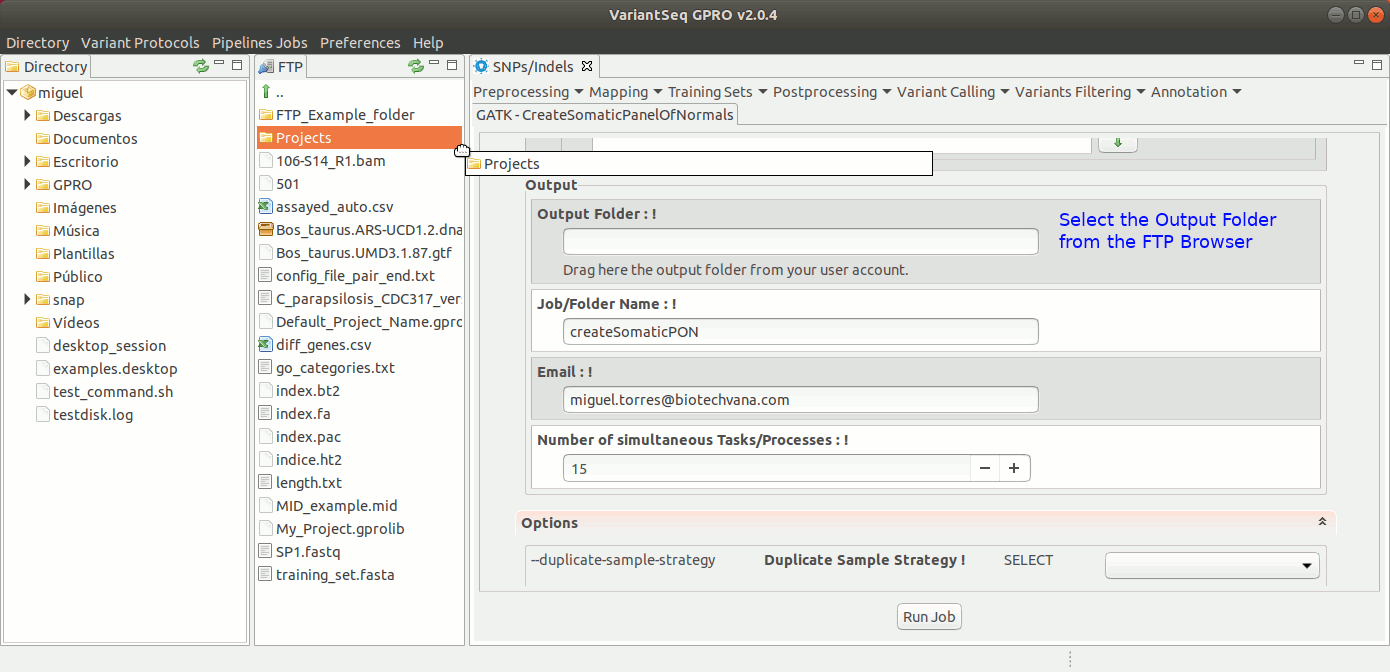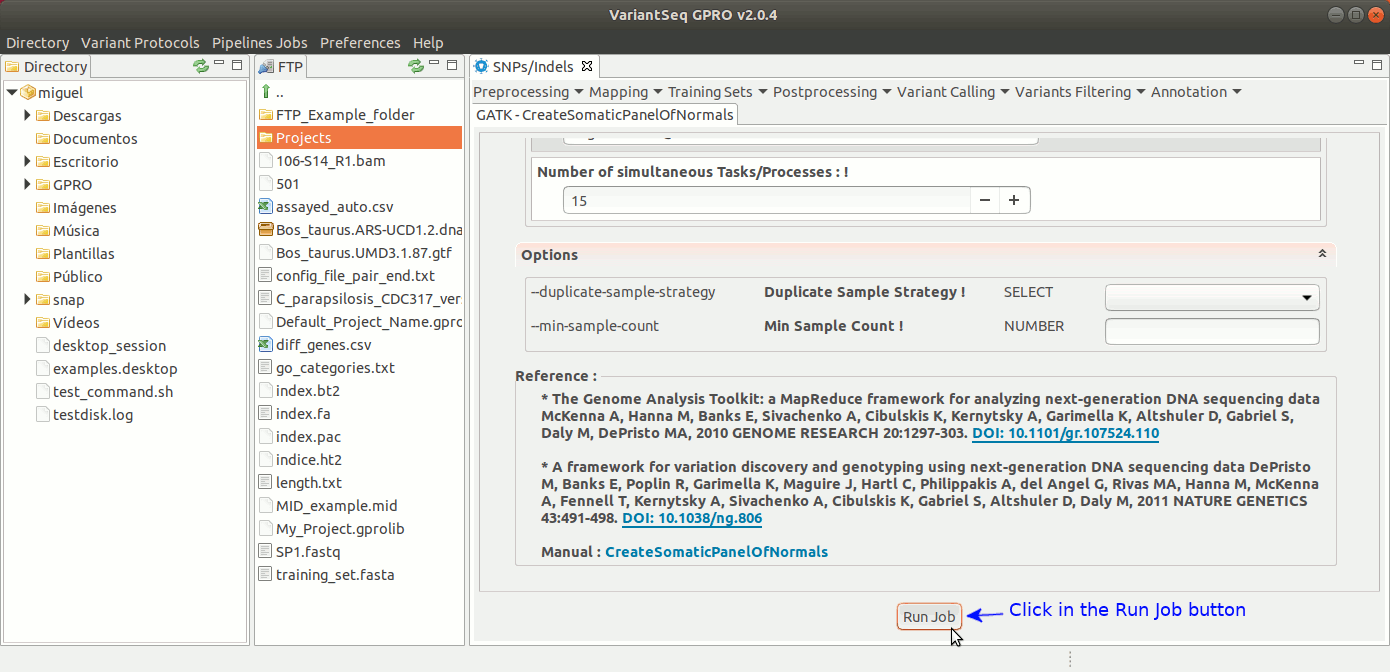
Figure 21: Using the GPRO interface for creating the PON.
Postprocessing helps to improve accuracy of the reads aligned to the reference in the bam file before the variant calling (Van der Auwera et al. 2013). The most typical post-processing tasks are: marking duplicates, local realignment around known indels, and base quality score recalibration (BQSR). VariantSeq implements different tools for preprocessing provided by GATK (McKenna et al, 2010, Picard tools (Wysoker et al 2011) and SAM tools (Li et al 2009).
- GATK Tools: BQSR
BQSR is required by GATK to correct systematic bias that affects the assignment of base quality scores by the sequencer. For more details see the GATK Forum
To execute GATK BQSR process, go to [ Postprocessing → GATK Tools → BQSR ] and follow Fig22.
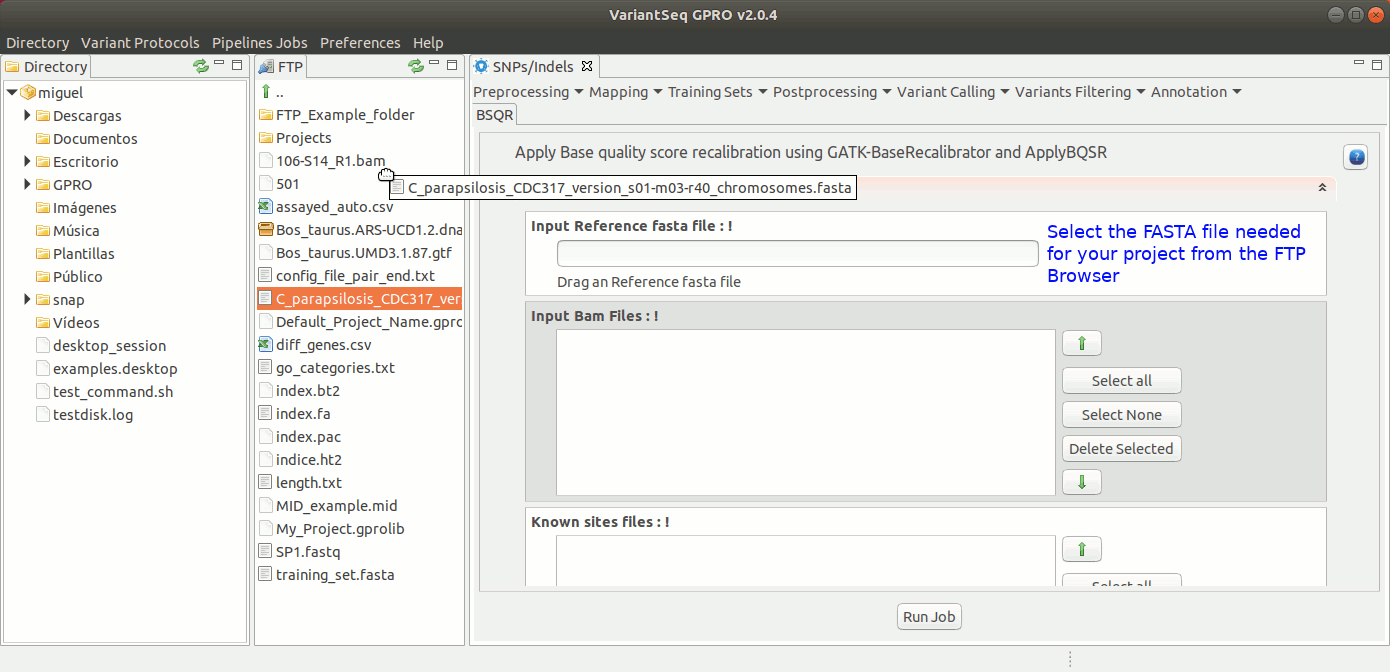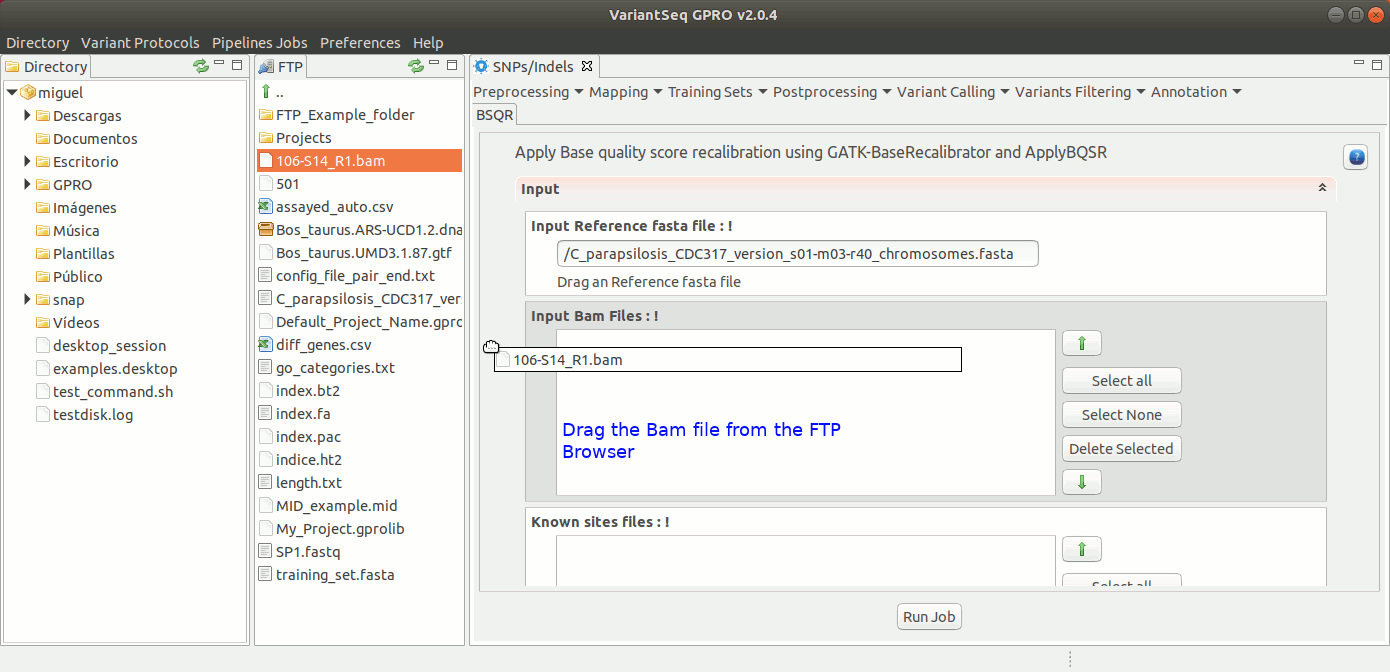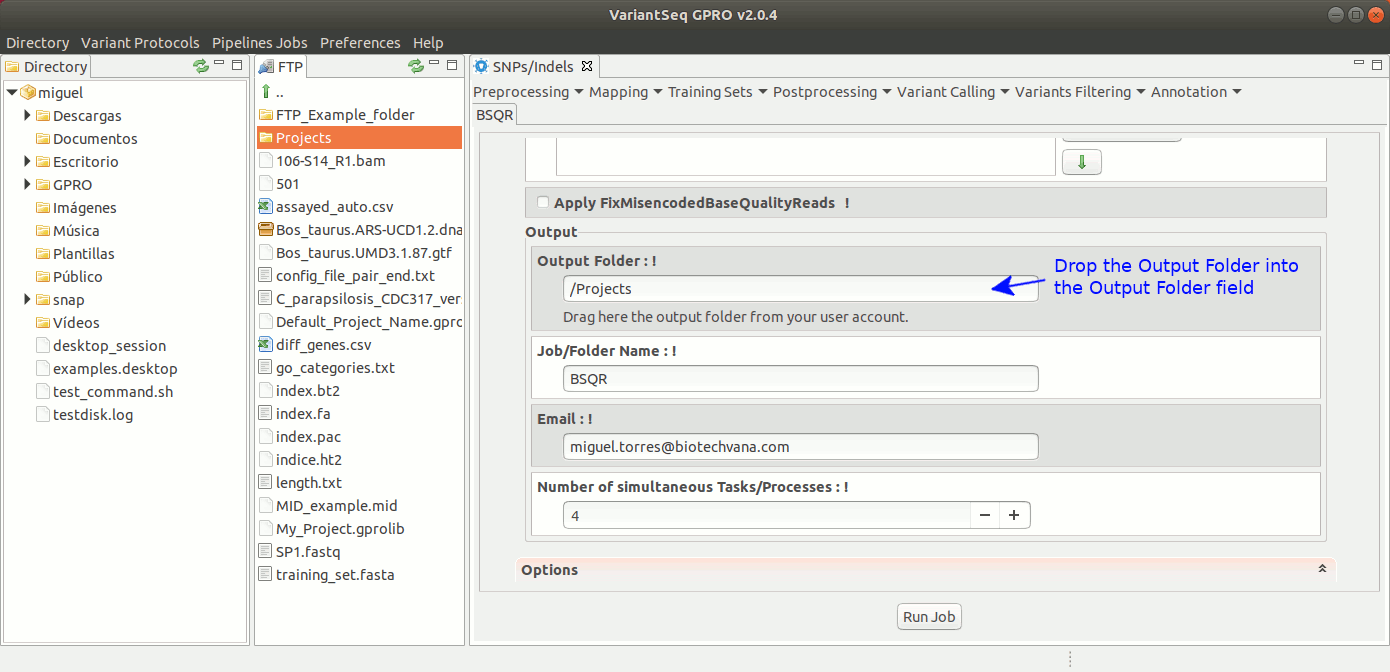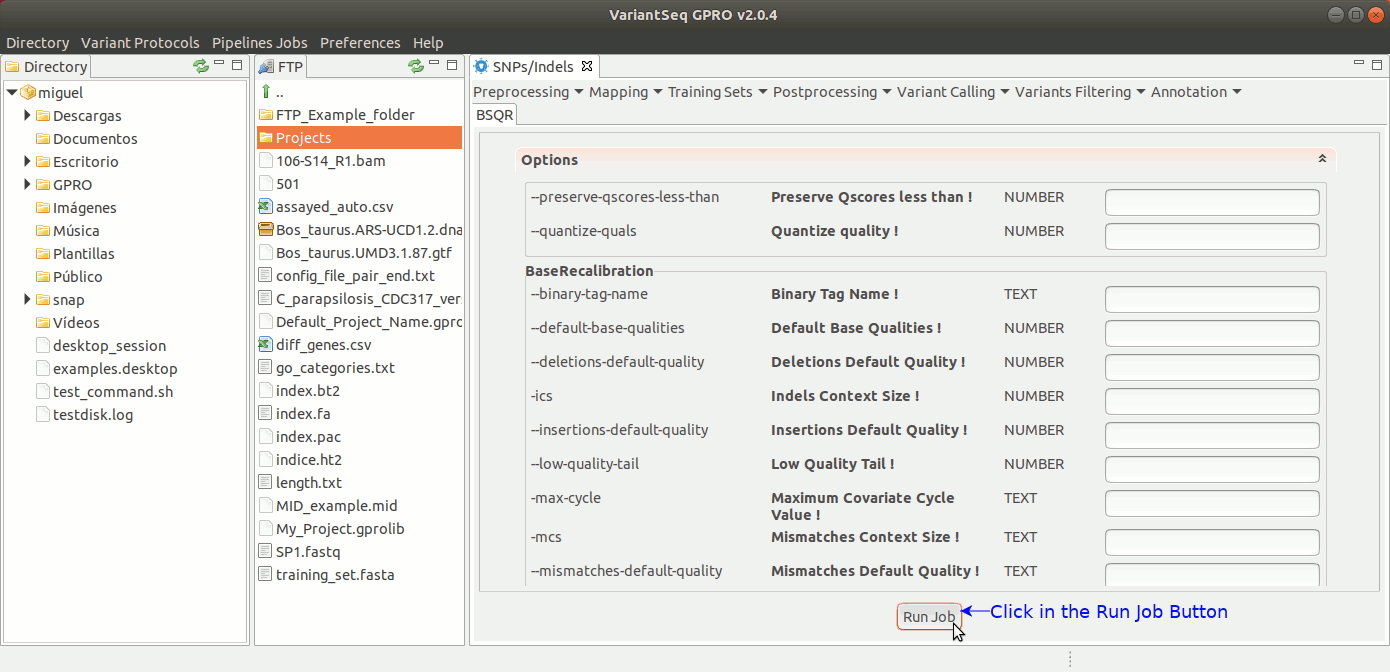
Figure 22: Using the GPRO interface for BQSR.
- GATK Tools: SplitNCigarReads
SplitNCigarReads is a tool of GATK specific for RNAseq data. It splits reads into exon segments (getting rid of Ns but maintaining grouping information) and hard-clips any overhanging sequences in the intronic regions. For more details see the GATK Forum
To run GATK SplitNCigarReads, go to [ Postprocessing → GATK Tools → SplitNCigarReads ] and follow Fig. 23.
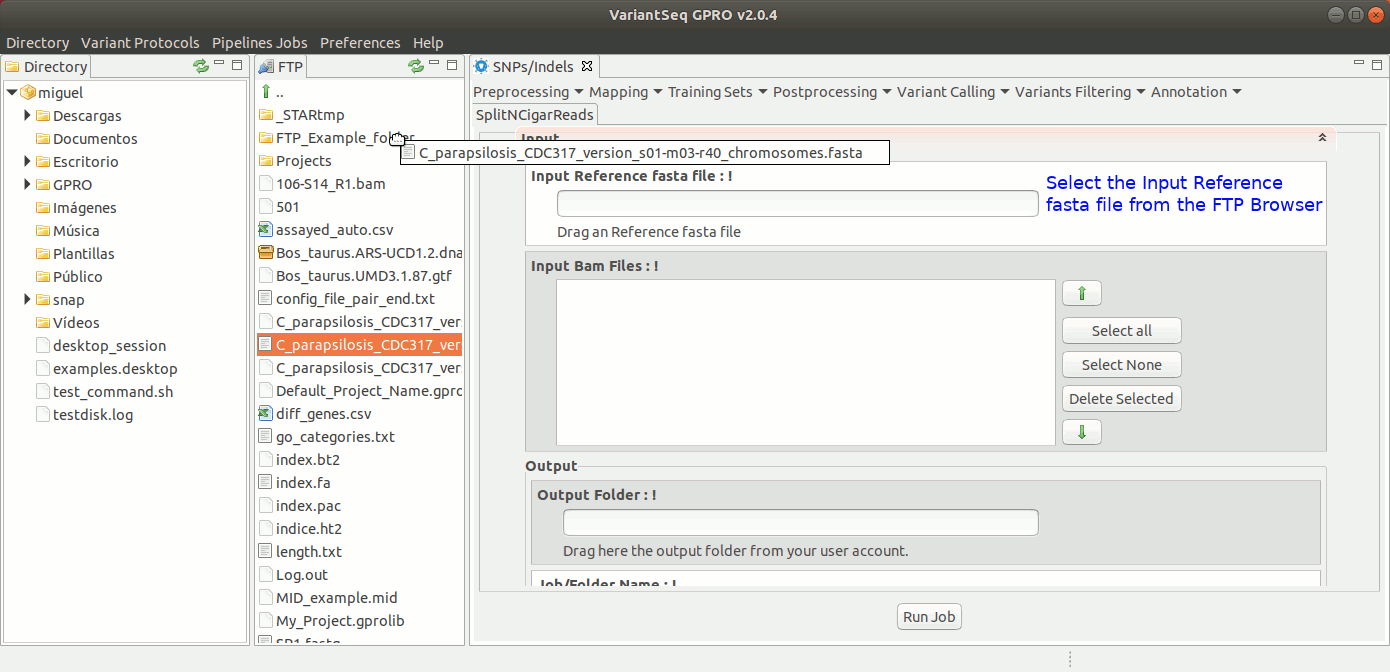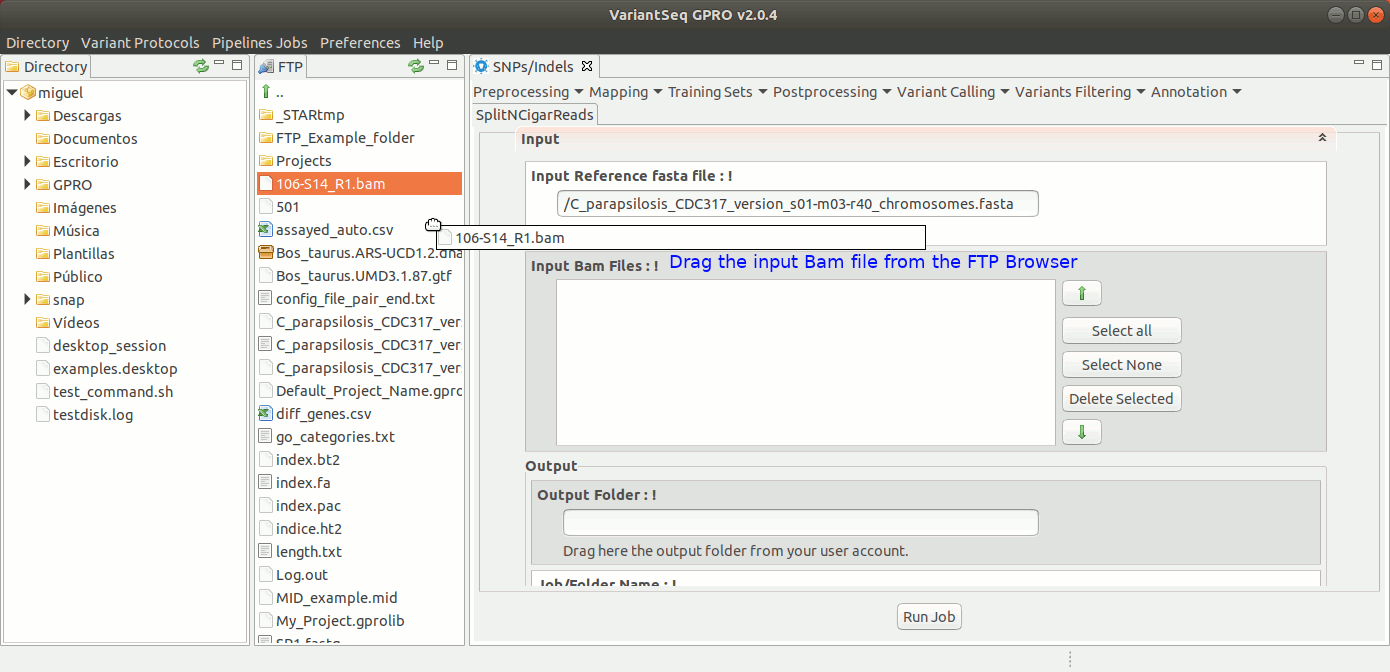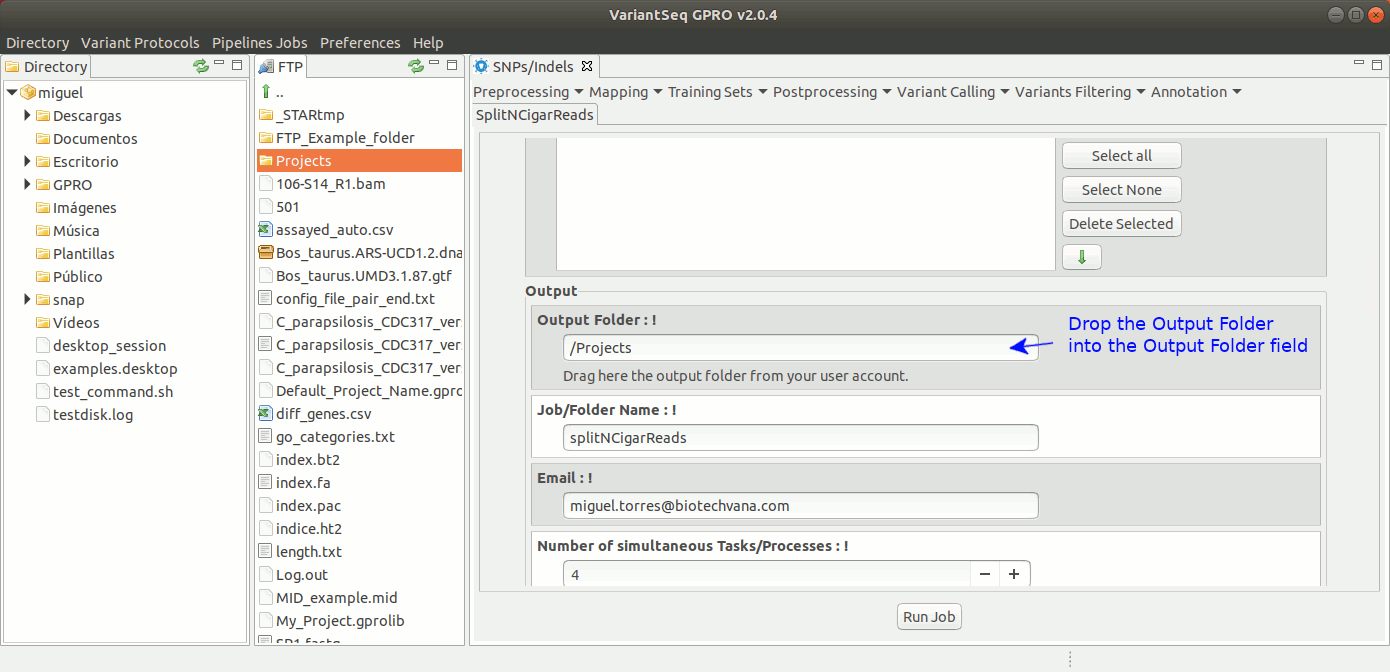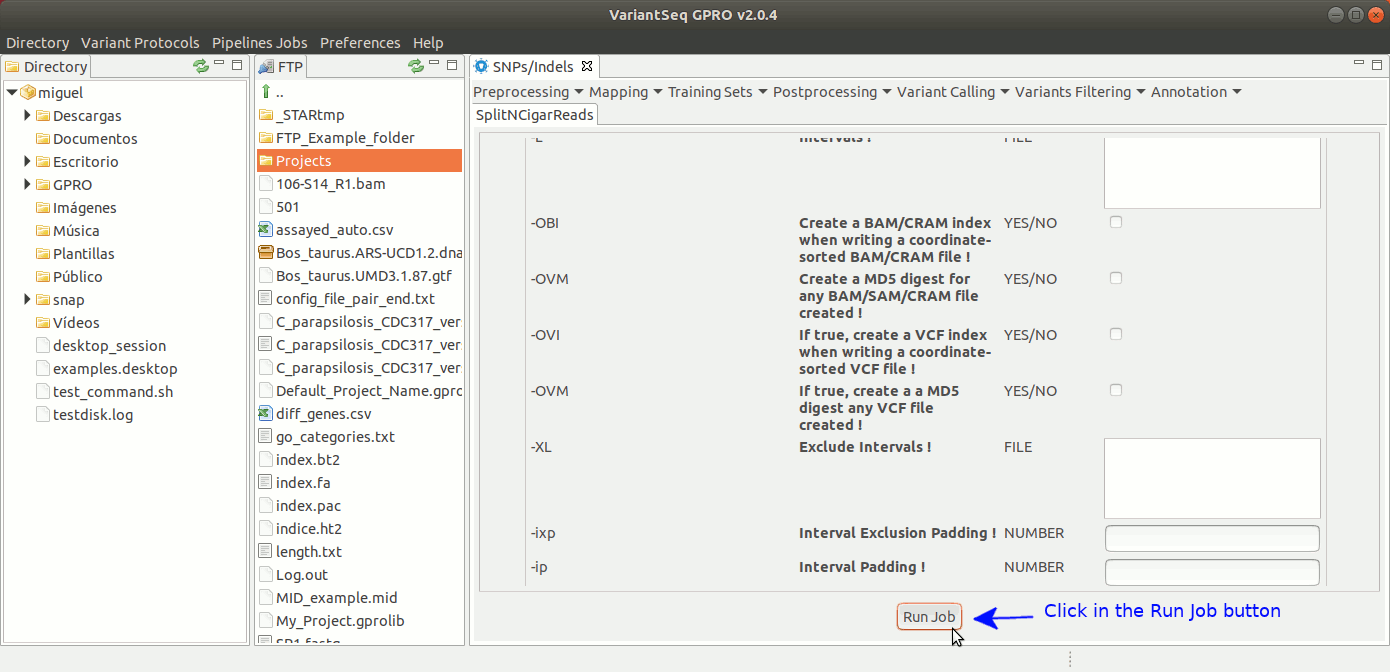
Figure 23: Using the GPRO interface for SplitNcigarReads
- GATK Tools: LeftAlignIndels
LeftAlignsIndels of GATK aligns indels in the left side of an alignment. While it is a common to place an indel at the left-most position, this is not always appropriate and indels may be placed at multiple positions. For more details see the GATK Forum
To run GATK LeftAlignIndels, go to [ Postprocessing → GATK Tools → LeftAlignIndels] and follow Fig. 24.
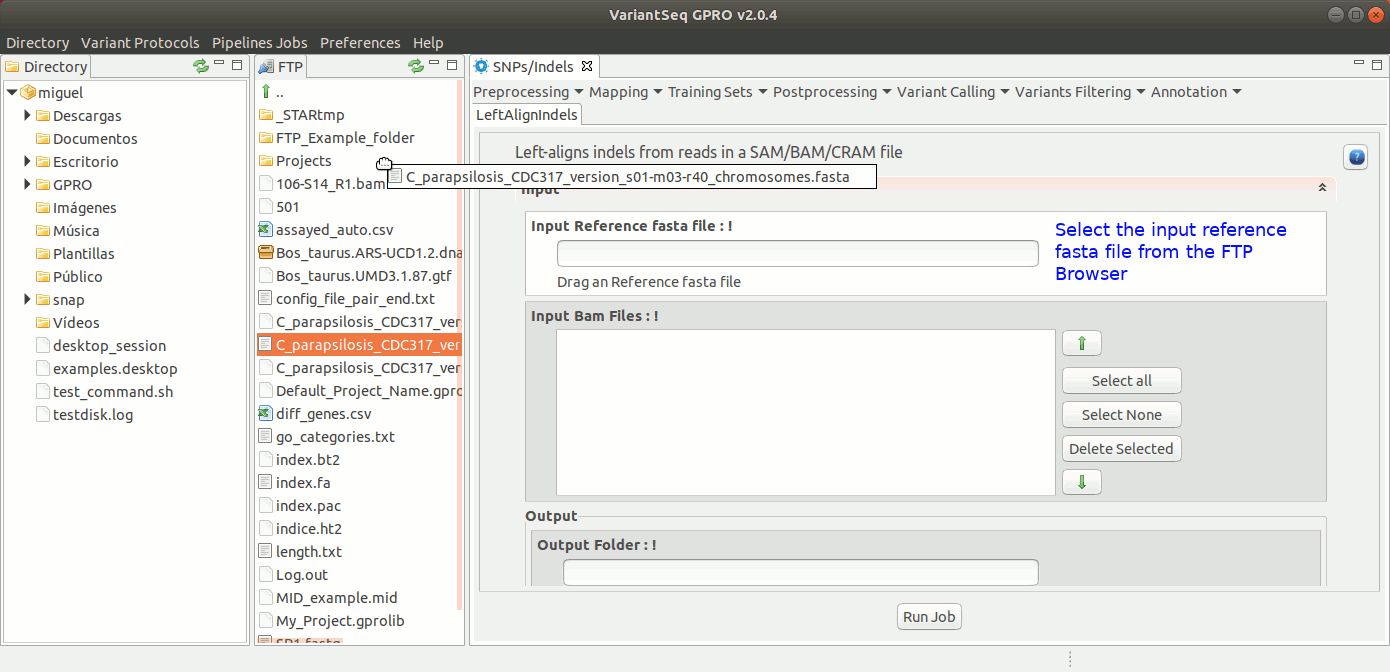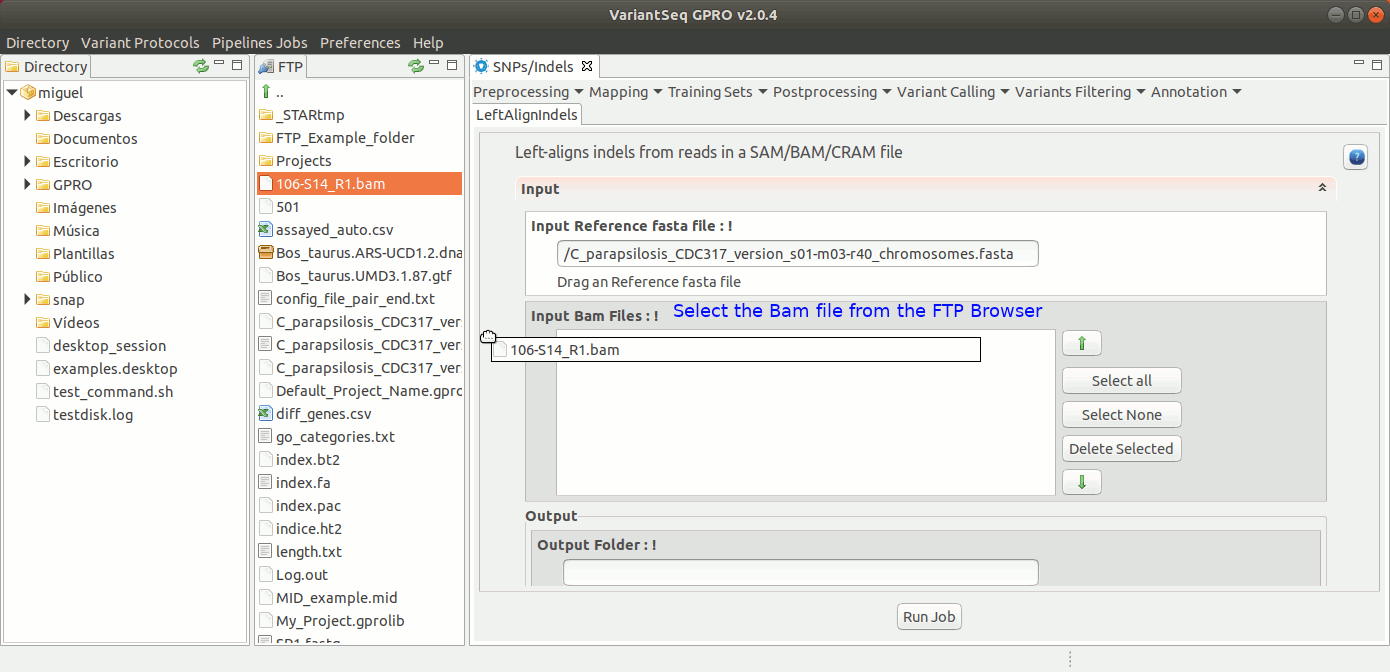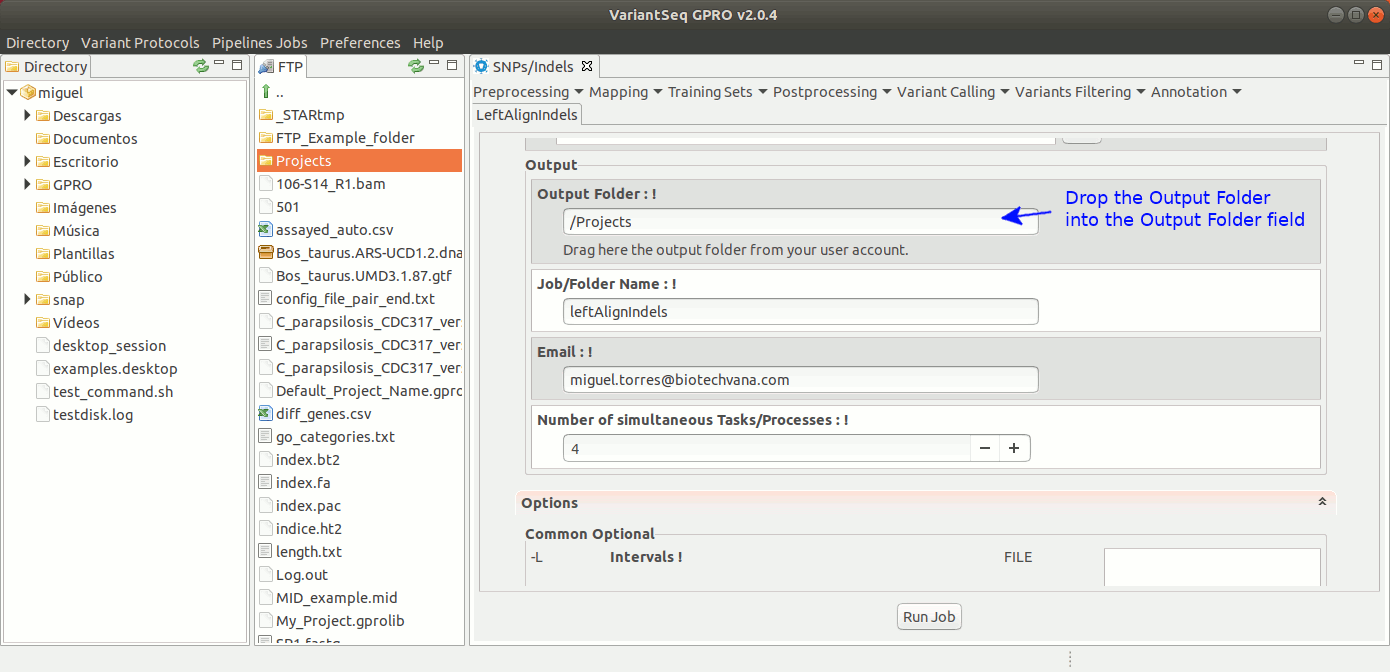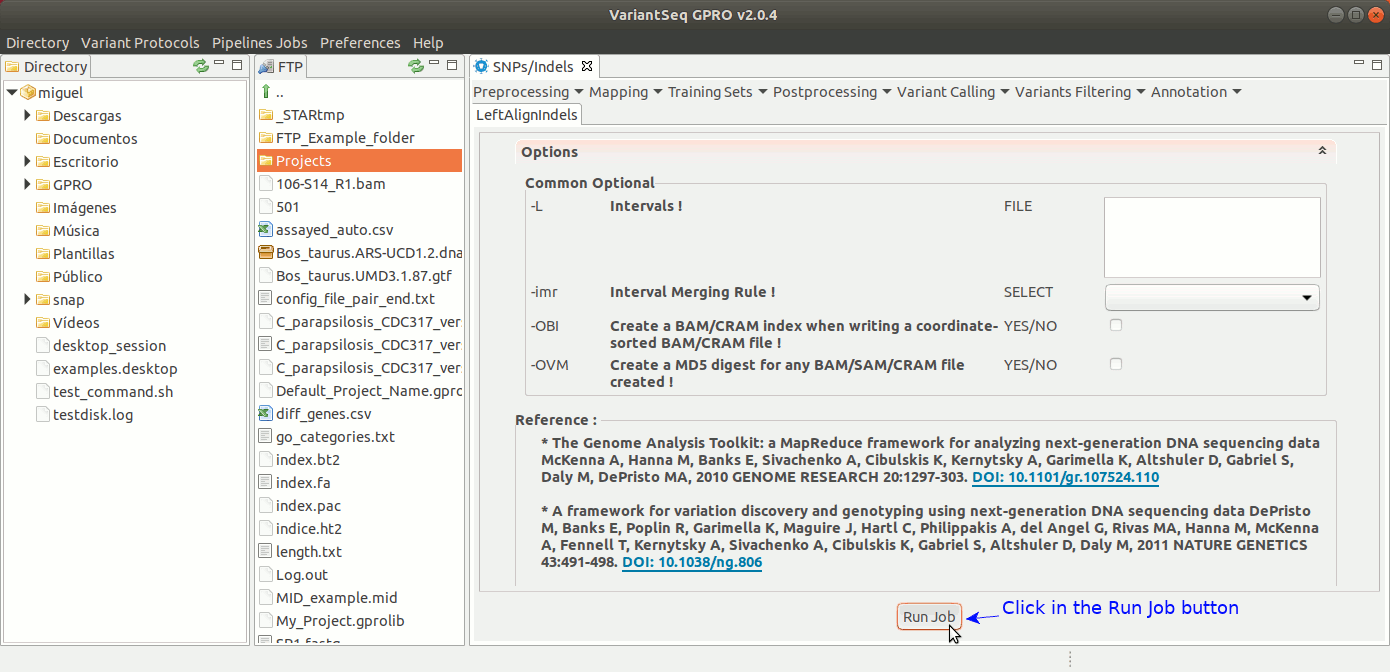
Figure 24: Using the GPRO interface for LeftAlignIndels,
- GATK Tools: IndelLocalRealignment
IndelLocalRealigment of GATK performs indel realignment using RealignerTargetCreator and IndelRealigner. This Function is not necessary for variant discovery if your plan is to use a caller that performs a haplotype assembly step, such as HaplotypeCaller or Mutec2. However, IndelLocalRealigment may be required when using legacy callers such as UnifiedGenotyper or the original Mutect. For more details see this thread at the GATK Forum
To run IndelLocalRealignment with VariantSeq, go to [ Postprocessing → GATK Tools → IndelLocalRealignment ] and follow Fig. 25.
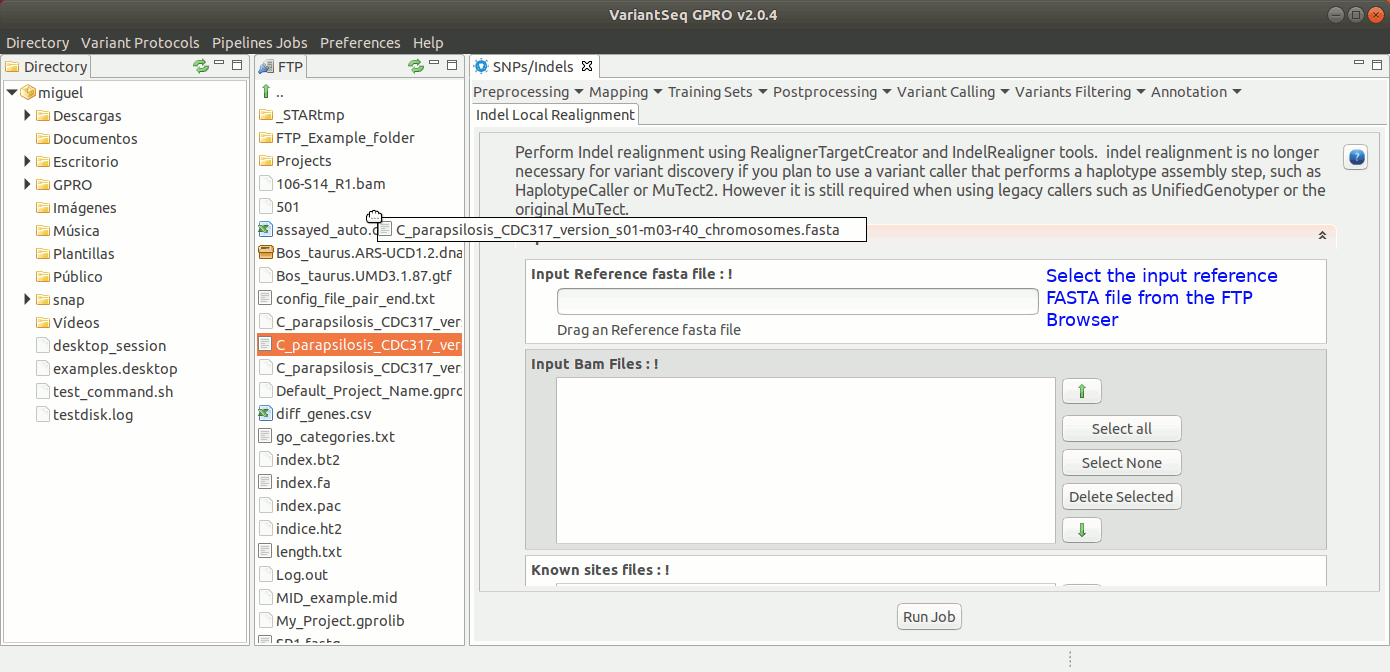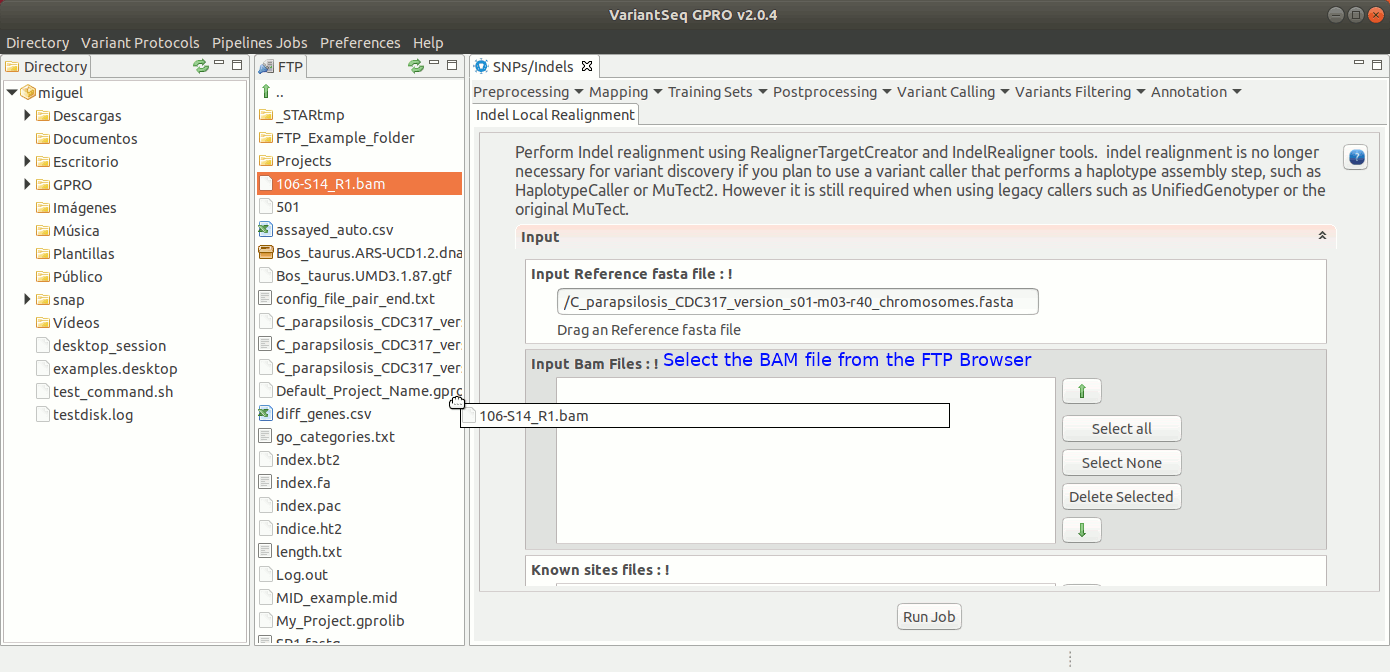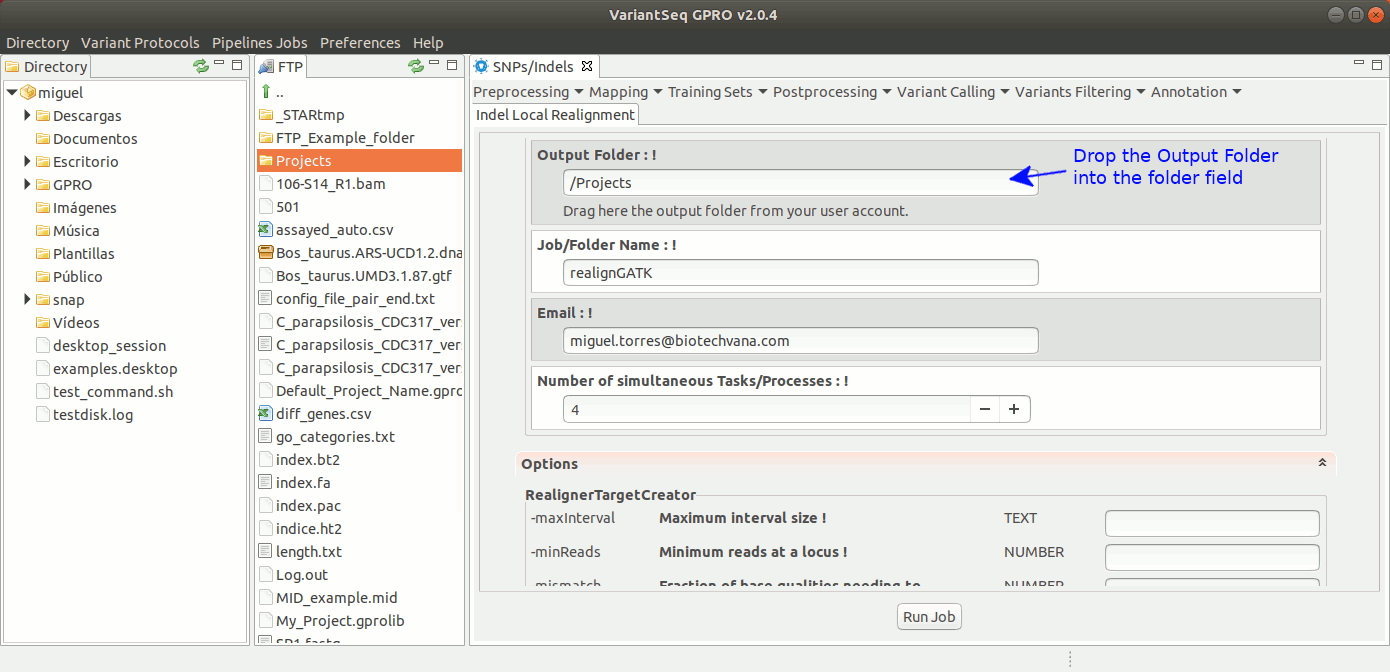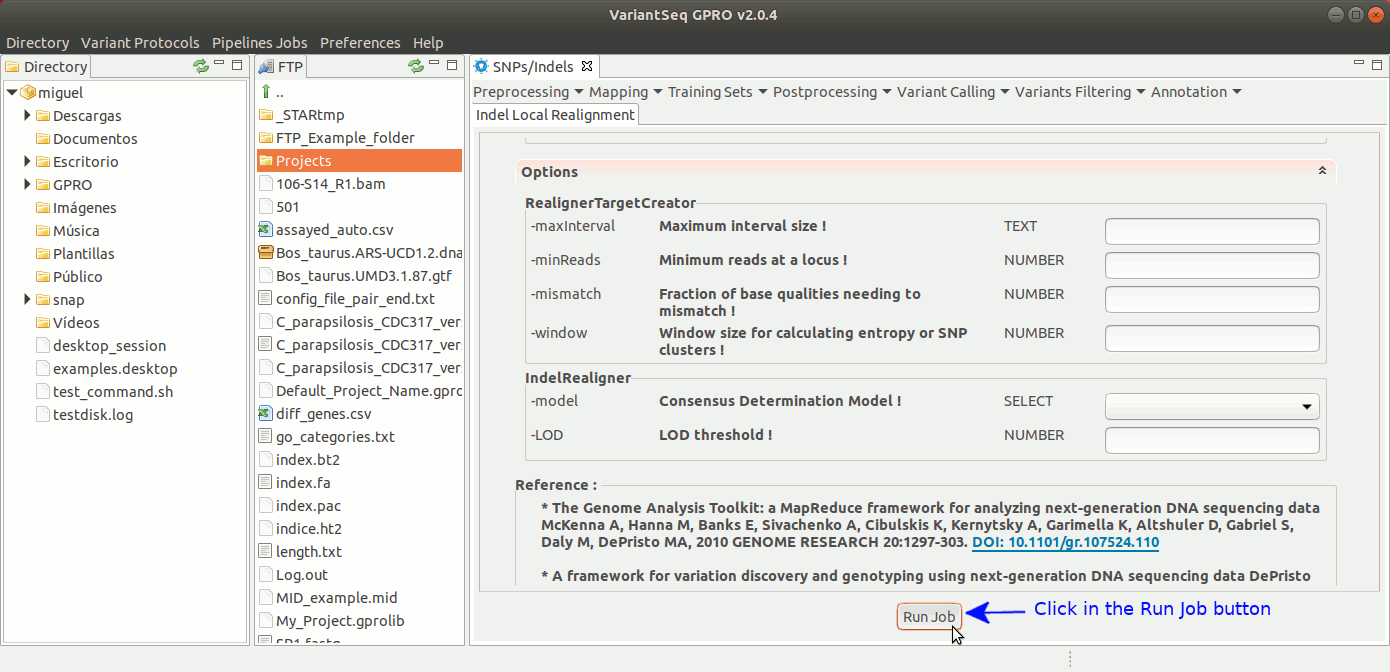
Figure 25: Using the GPRO interface for IndelLocalRealignment,
- Picard Tools: AddOrReplaceReadGroups
AddOrReplaceReadGroups is a tool withein Picard to combin all the reads of a file to a new read-group. The tool accepts INPUT BAM and SAM files. For more details see the GATK Forum
To execute AddOrReplaceReadGroups, go to [ Postprocessing → Picard Tools→AddOrReplaceReadGroups ] and follow Fig. 26.
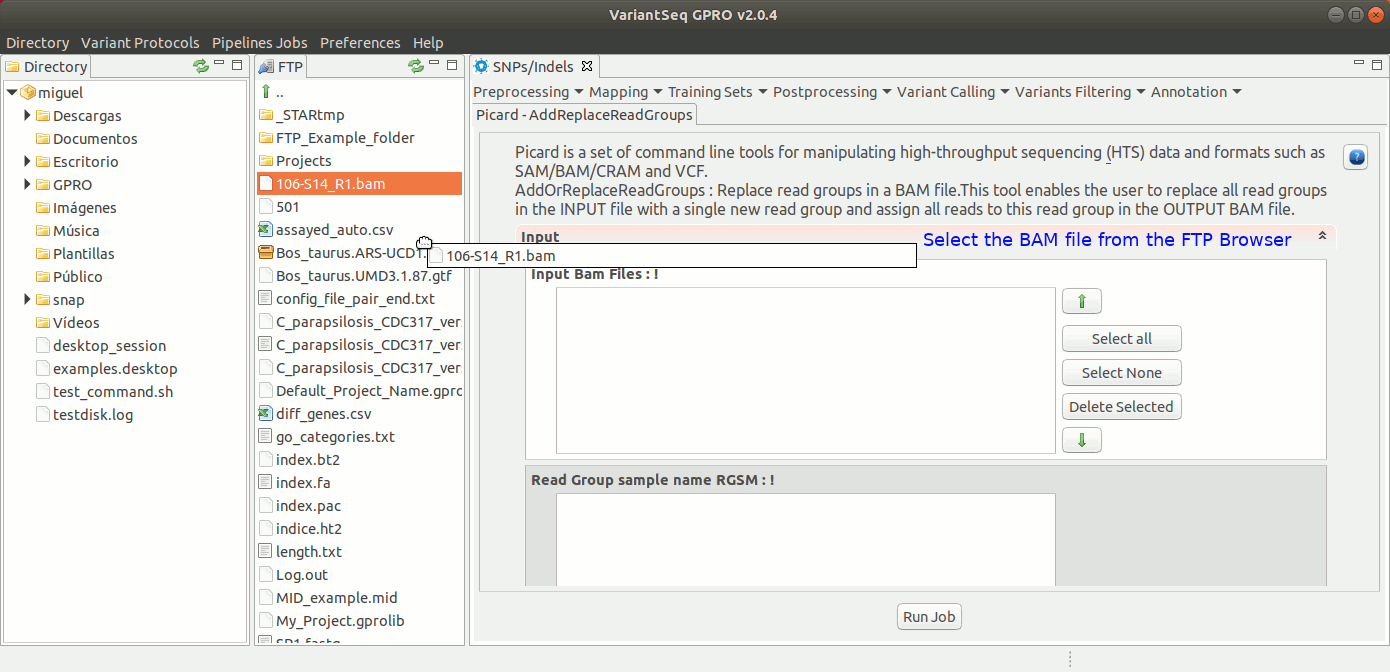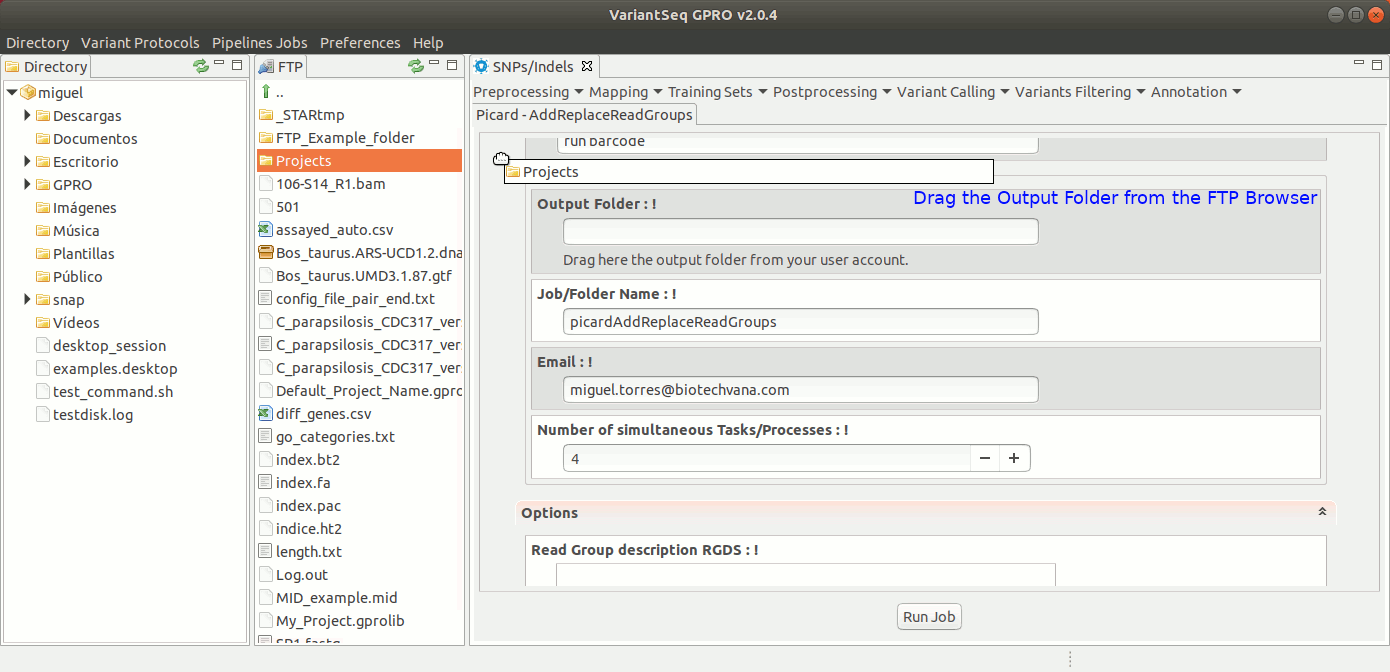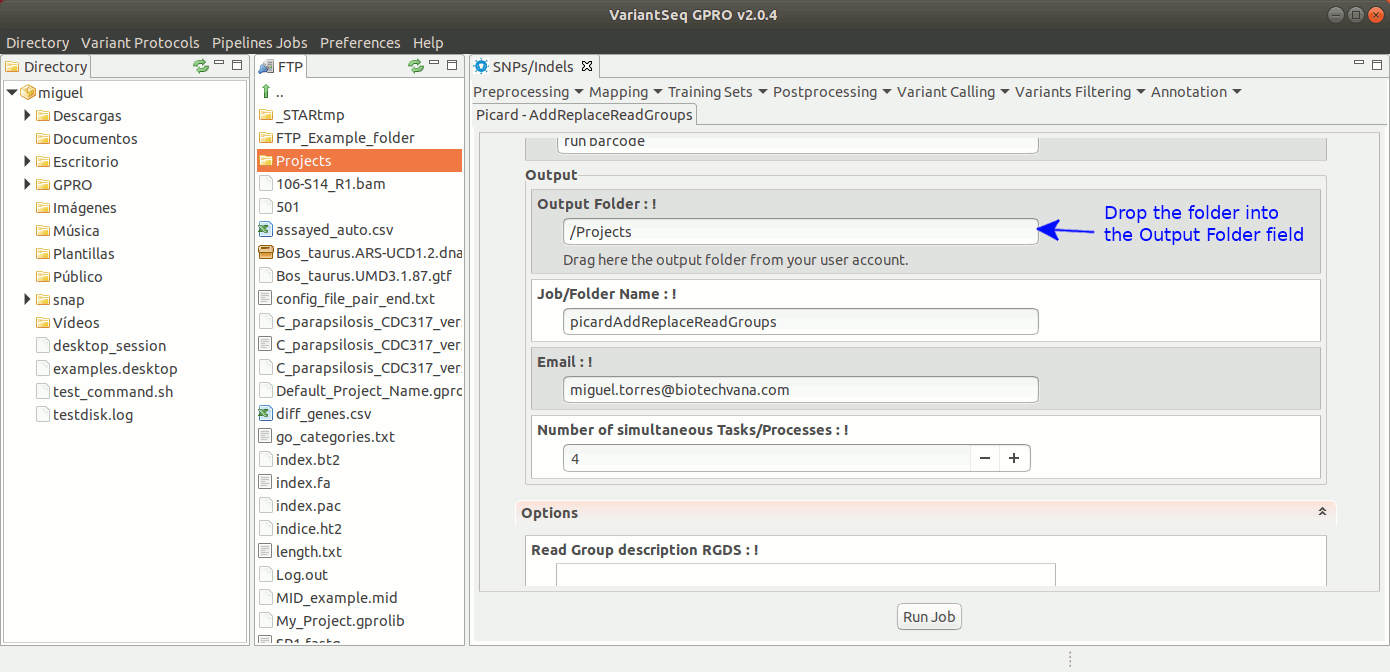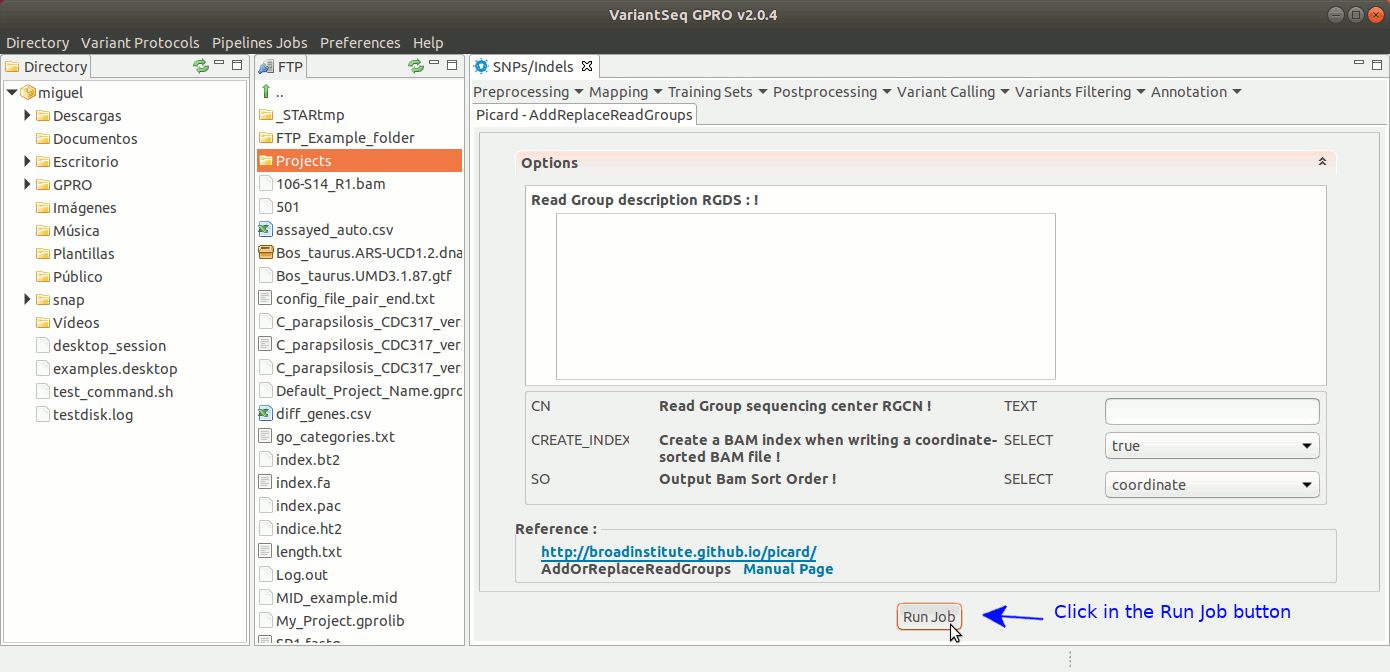
Figure 26: Using the GPRO interface for AddOrReplaceReadGroups,
- Picard Tools: CreateSequenceDictionary
CreateSequenceDictionary of Picard creates a sequence dictionary file (with the extension .dict) from a reference fasta file. The sequence dictionary is required by several steps and tools of the variant analysis. For more details see the : GATK Forum
To execute CreateSequenceDictionary, go to [ Postprocessing →Picard Tools → CreateSequenceDictionary ] and follow Fig. 27.
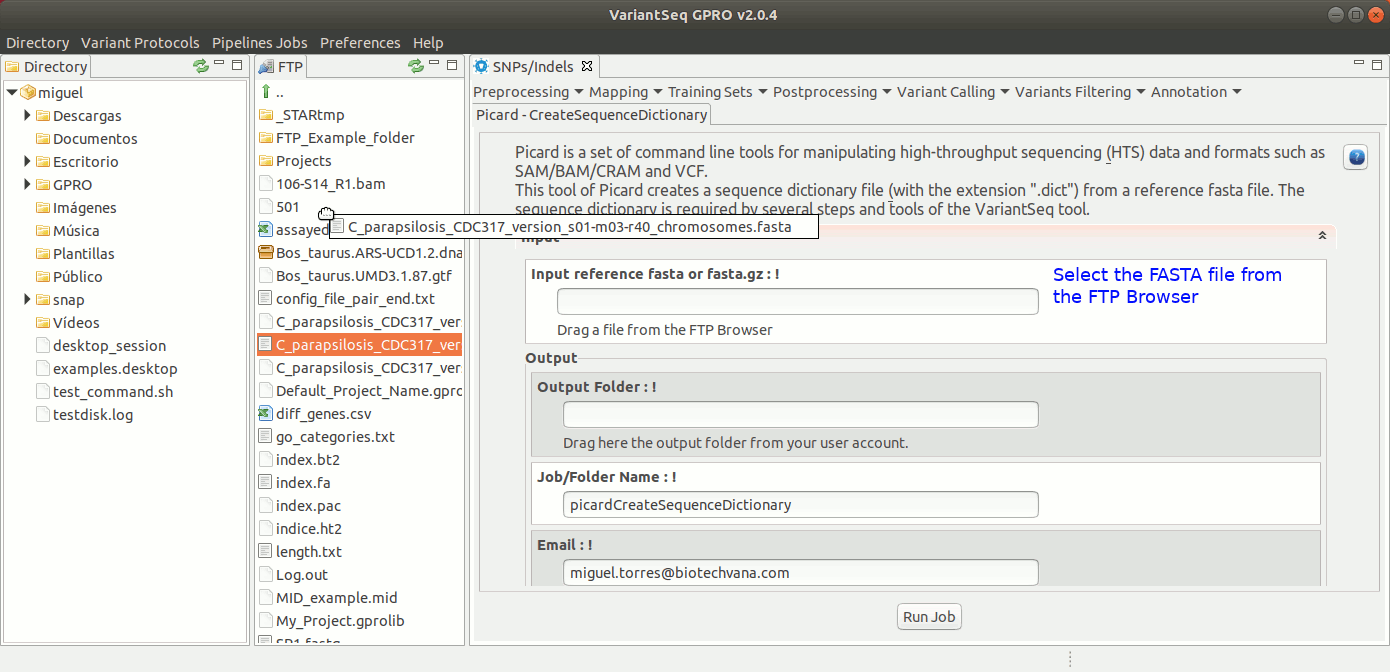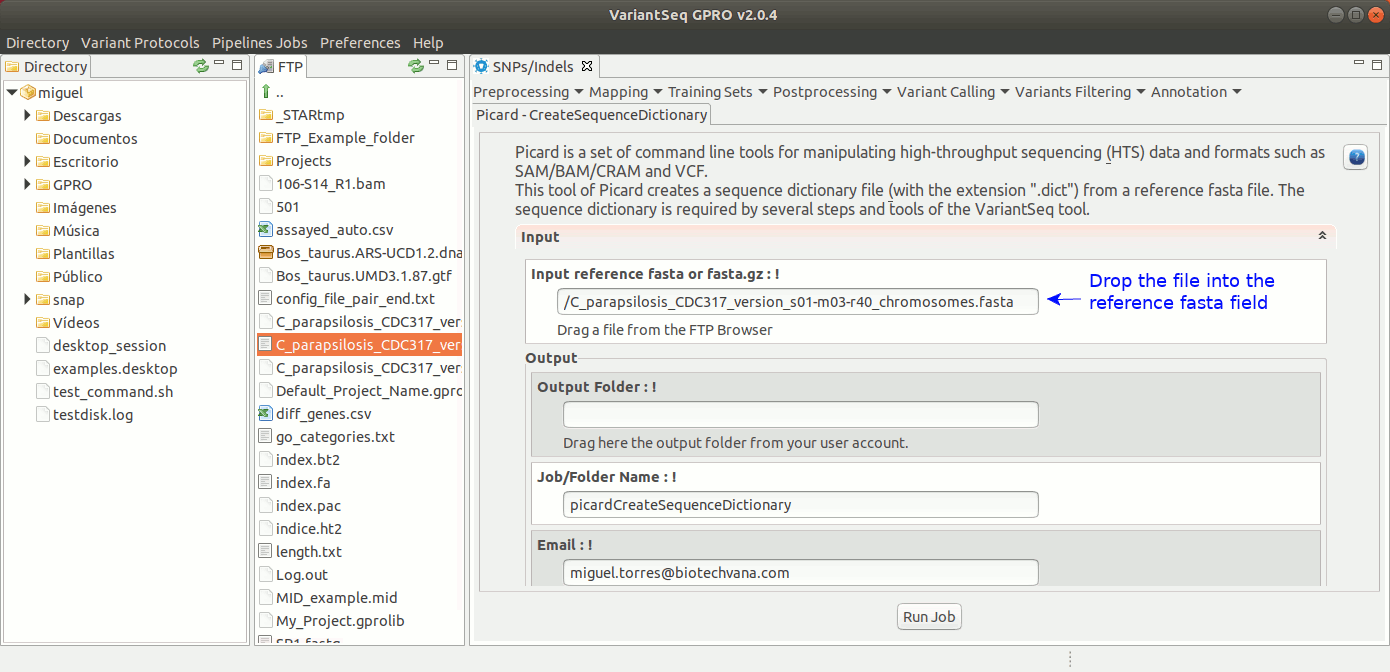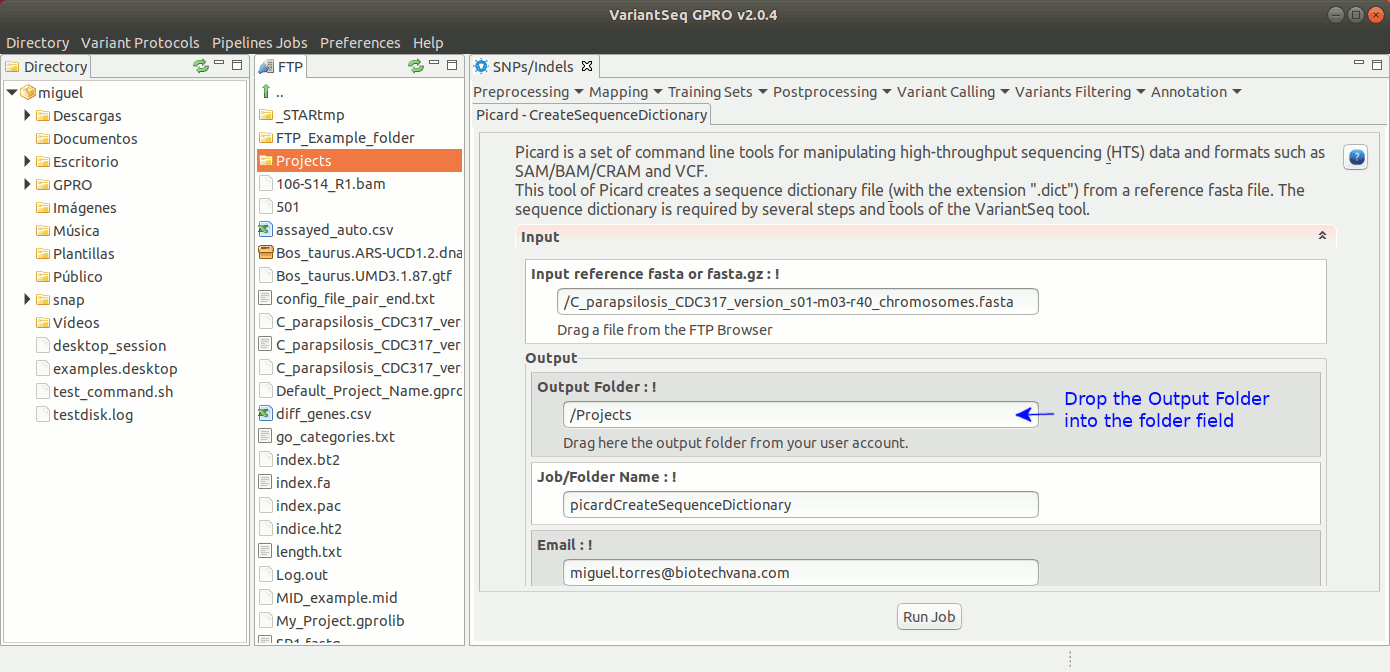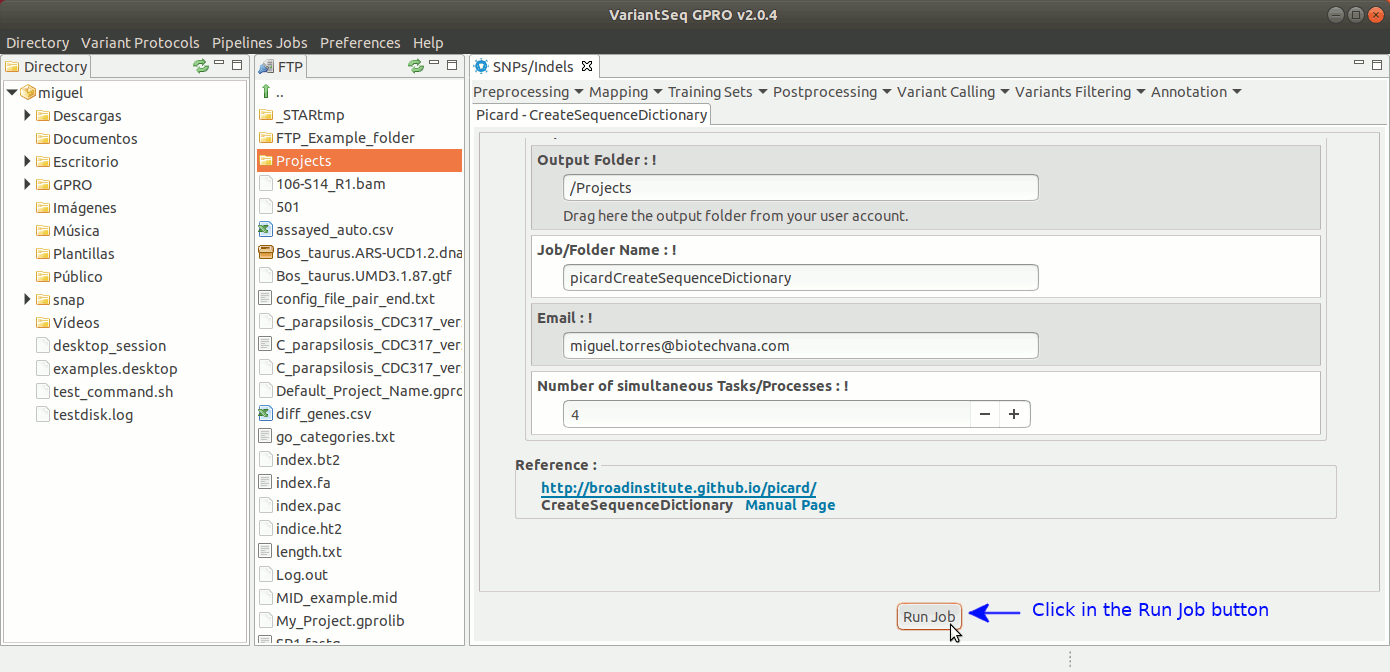
Figure 27: Using the GPRO interface for CreateSequenceDictionary,
- Picard Tools: MarkDuplicates
MarkDuplicates of Picard locates and tags duplicate reads in a BAM or SAM file. For more details see the GATK Forum
To execute MarkDuplicates, go to [ Postprocessing →Picard Tools → MarkDuplicates ] and follow Fig. 28.
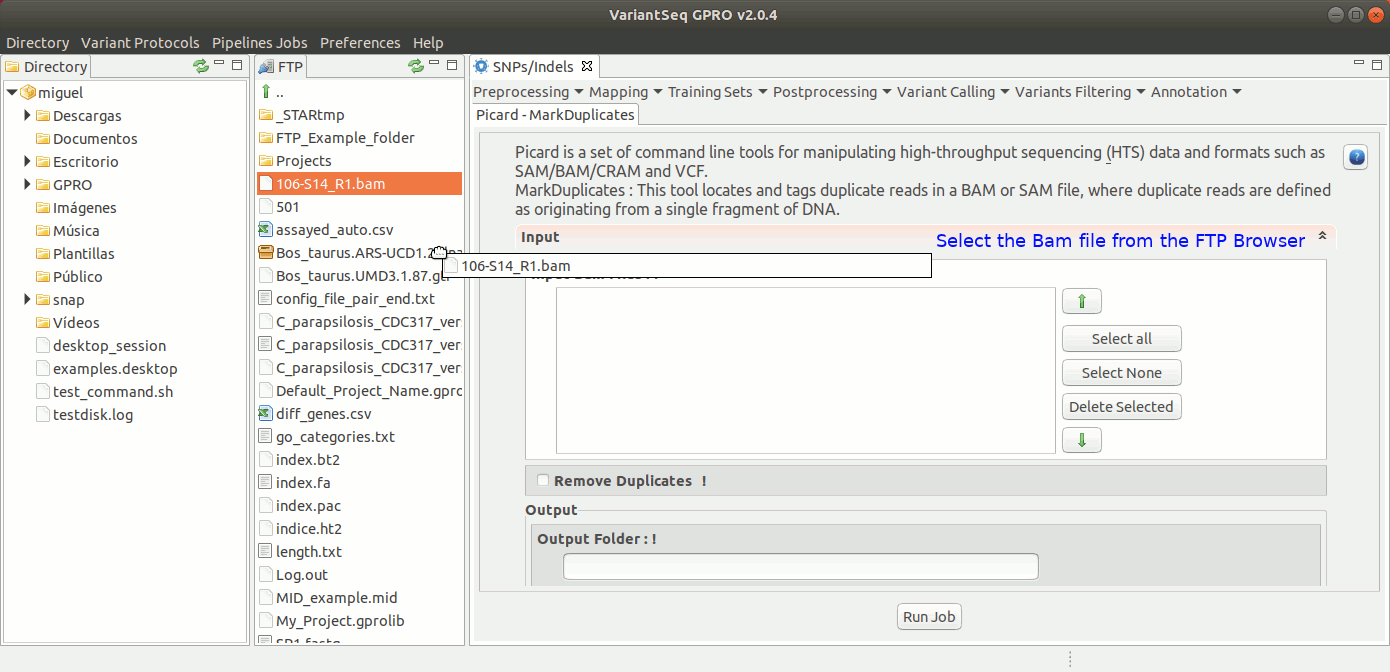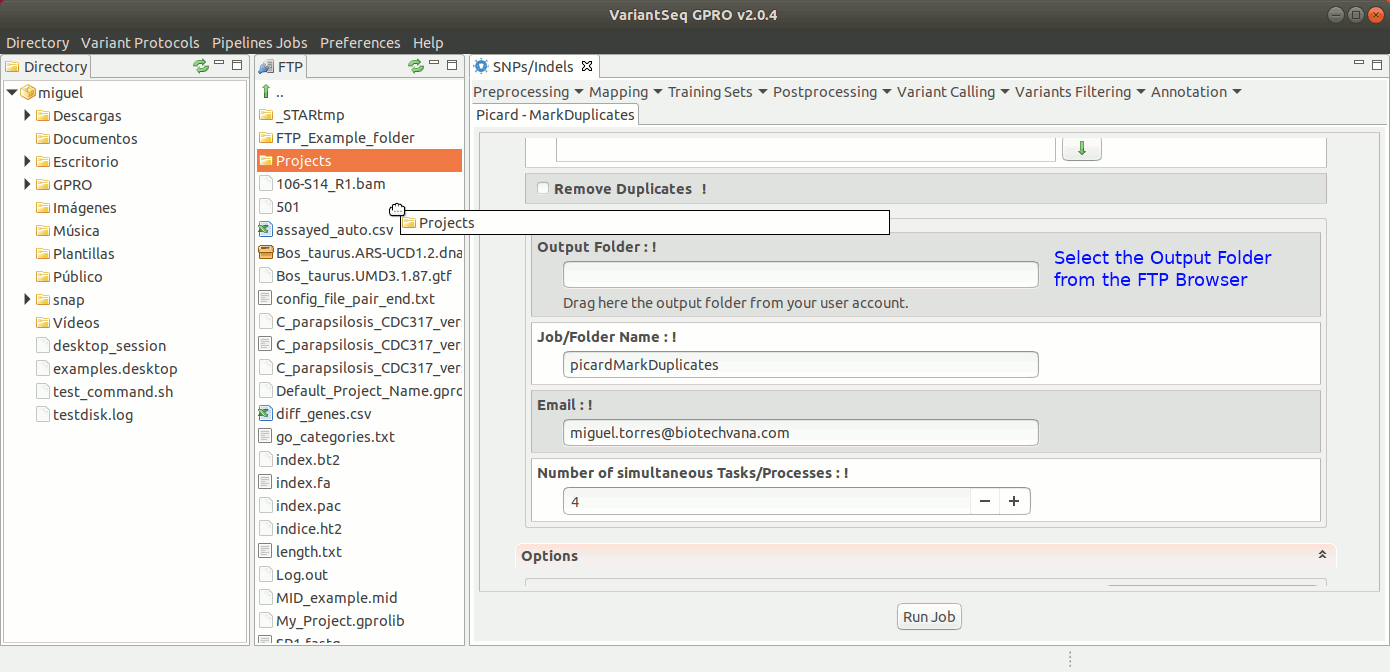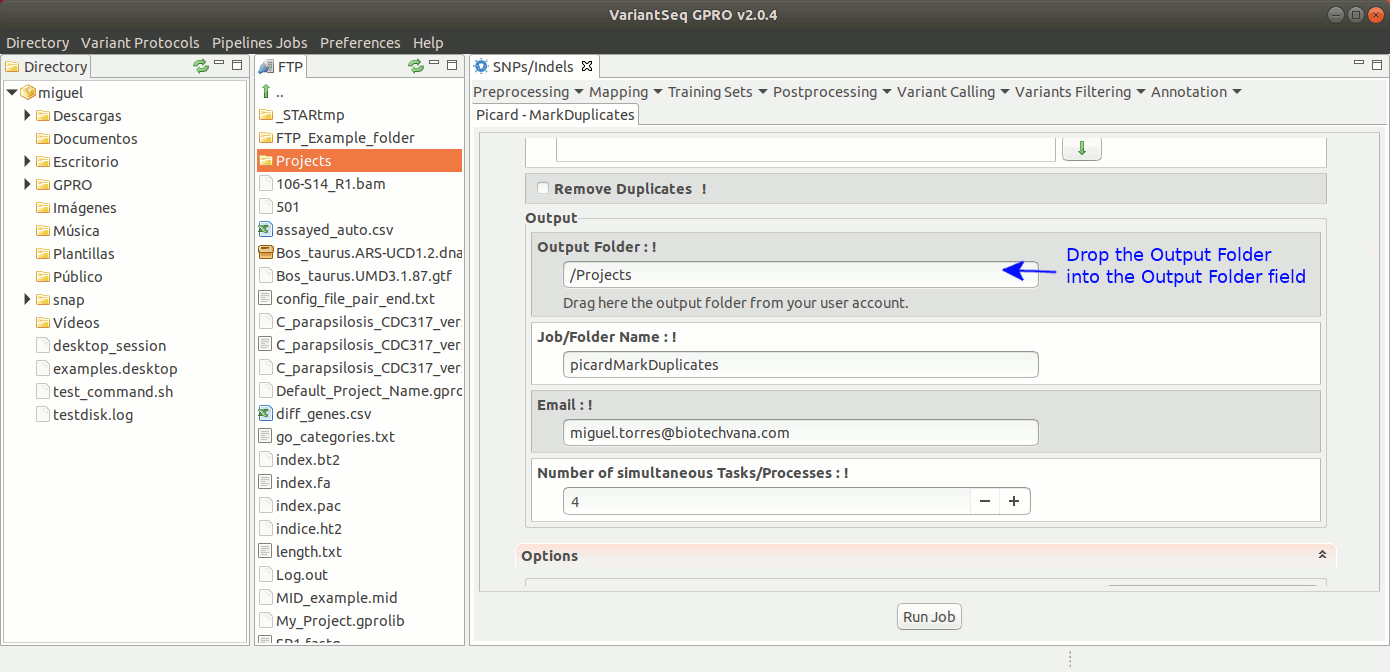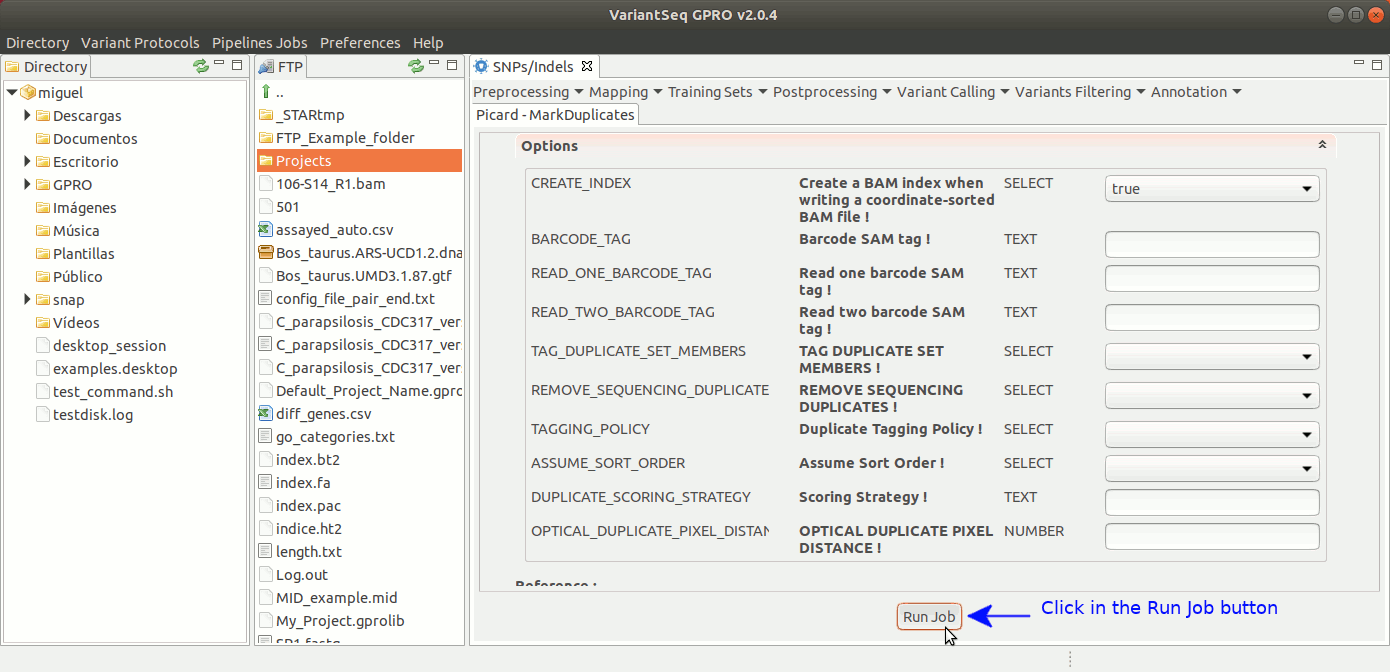
Figure 28: Using the GPRO interface for MarkDuplicates,
- SAM Tools: Index
The Index utility of SamTools creates a .bai or a .csi Index file from a bam file. For more details see the samtools manual at http://samtools.sourceforge.net
To run samtools index with VariantSeq, go to [ Postprocessing → SamTools → Index ] and follow Fig. 29.
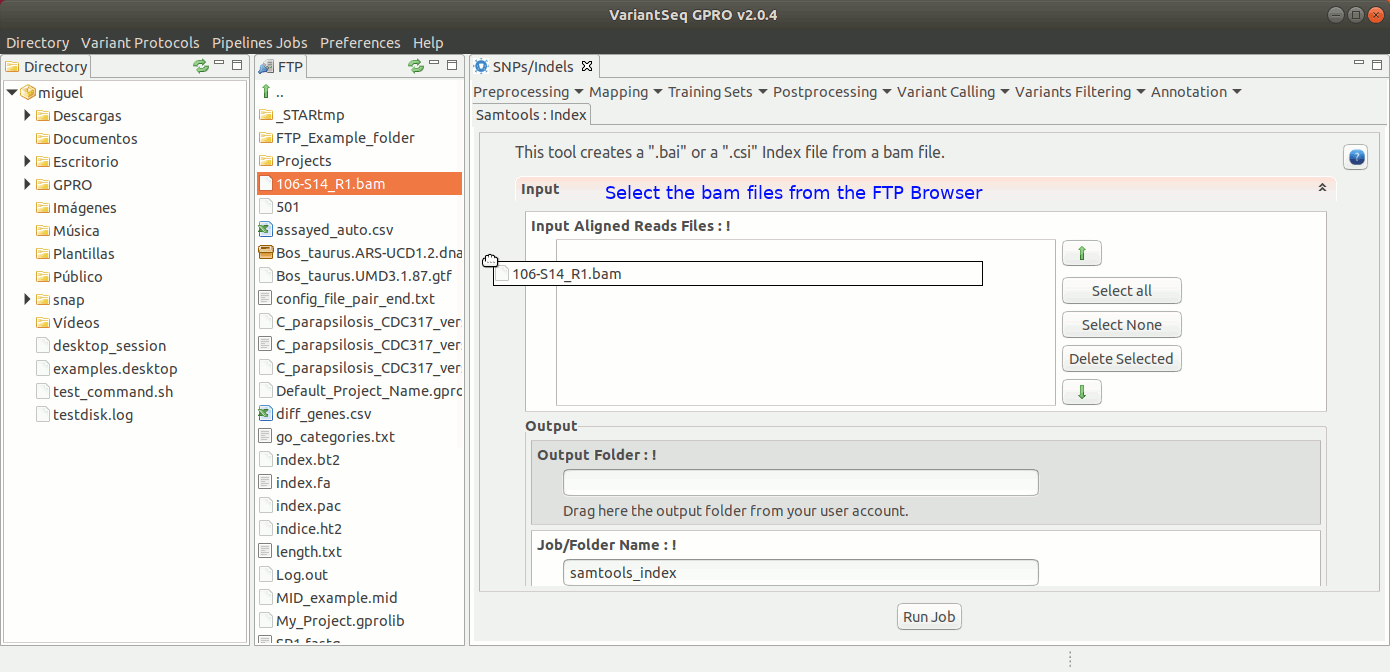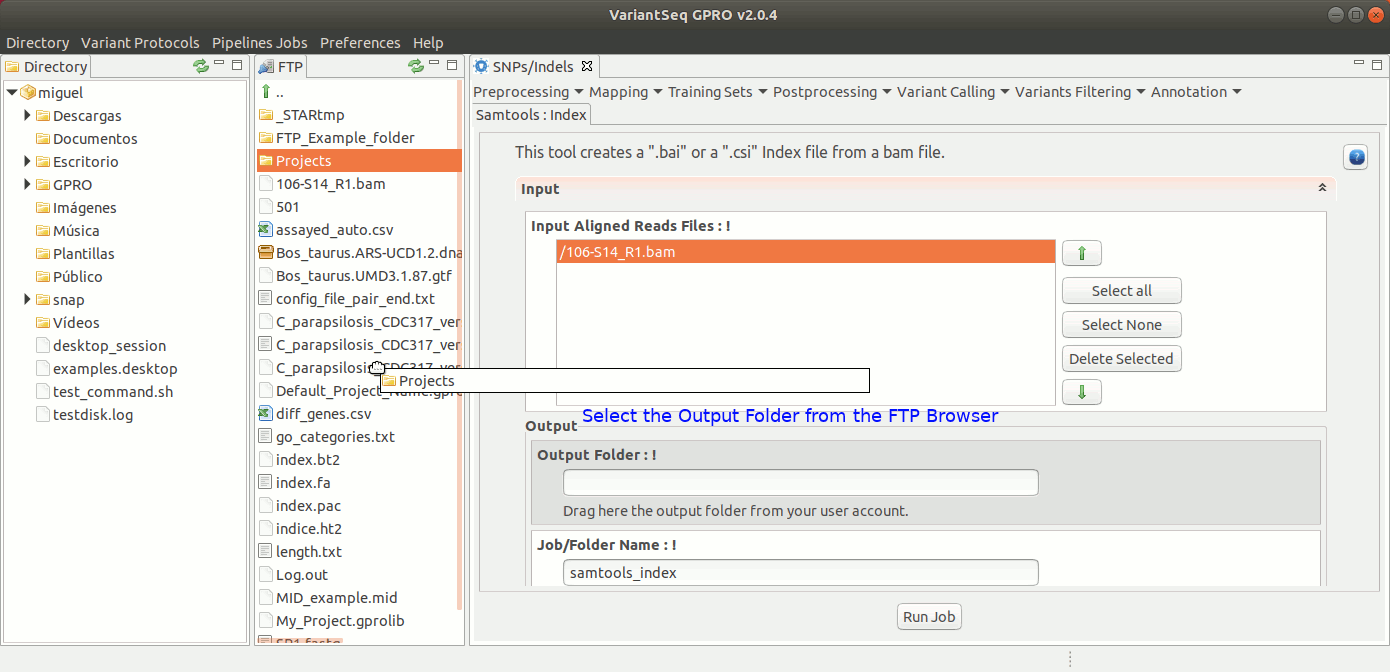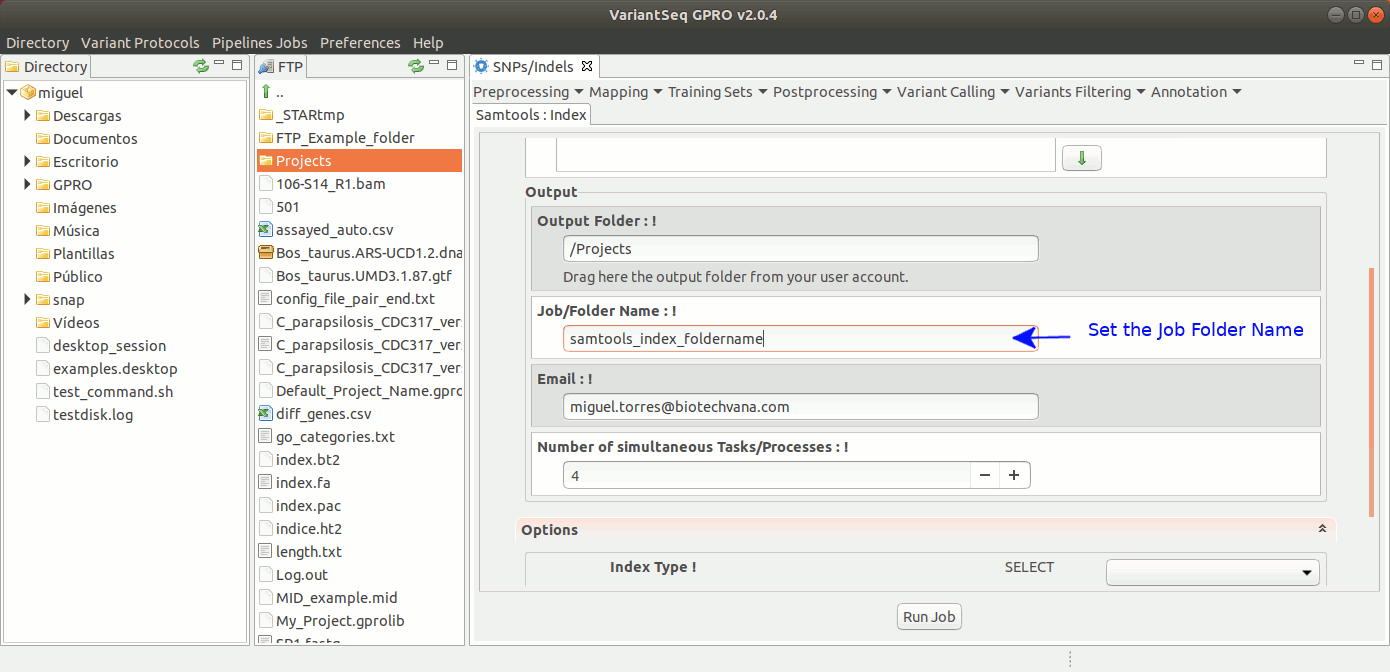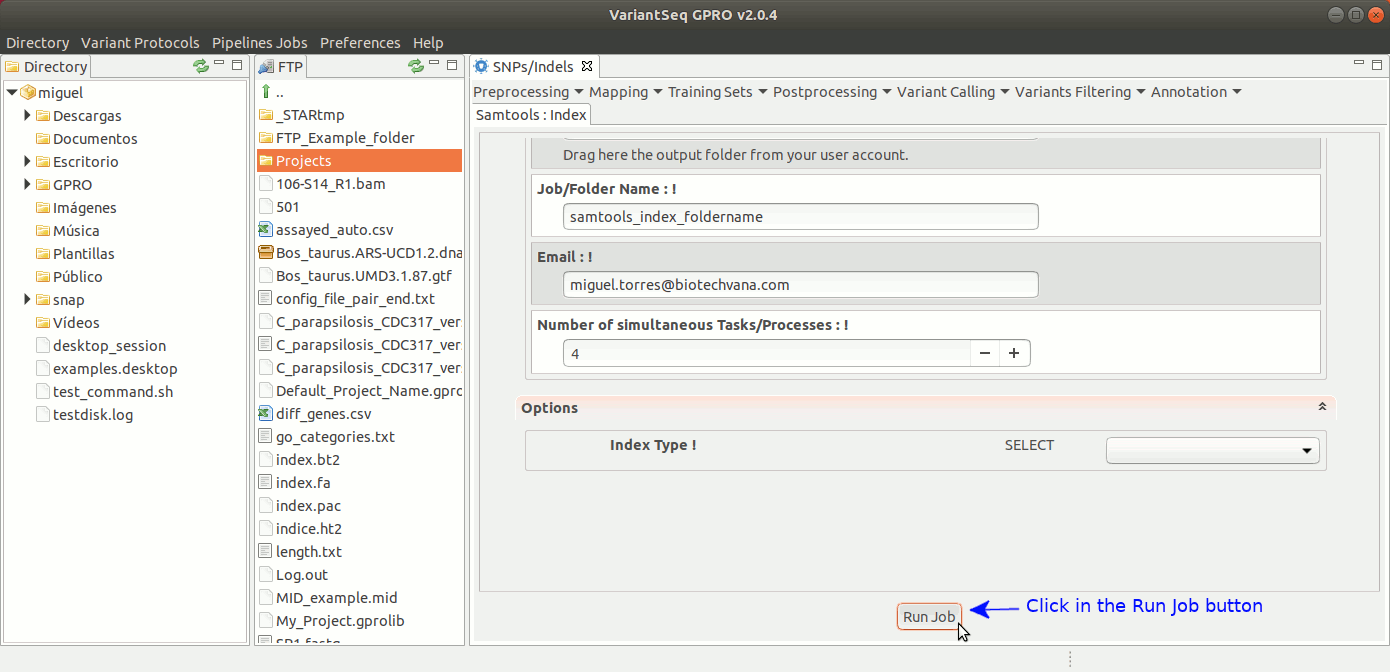
Figure 29: Using the GPRO interface for Samtools Index.
- SAM Tools: Sort
Sort is a tool of SamTools that sorts alignments in bam files either by leftmost coordinates or by read name when -n is used. If necessary, an appropriate @HD-SO sort order header tag will be added or an existing one updated. For more details see the samtools manual at http://samtools.sourceforge.net
To run Samtools Sort with VariantSeq, go to [ Postprocessing → Samtools → Sort ] and follow Fig. 30.
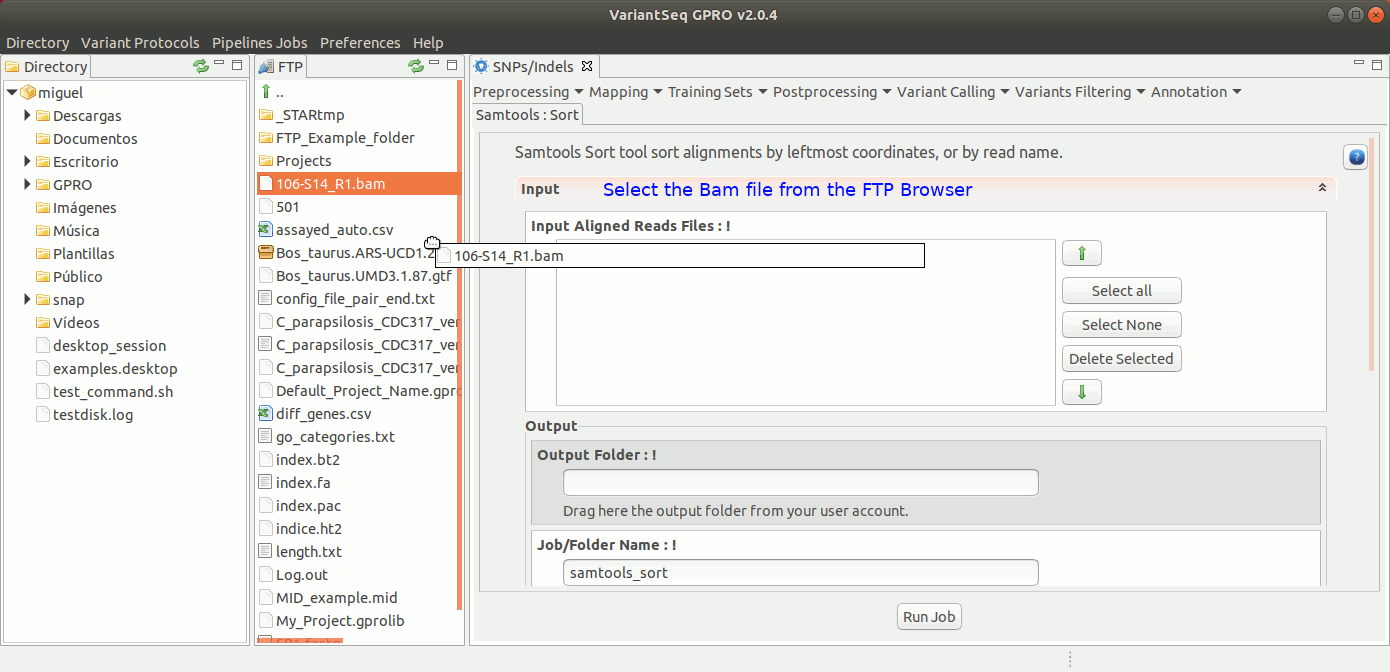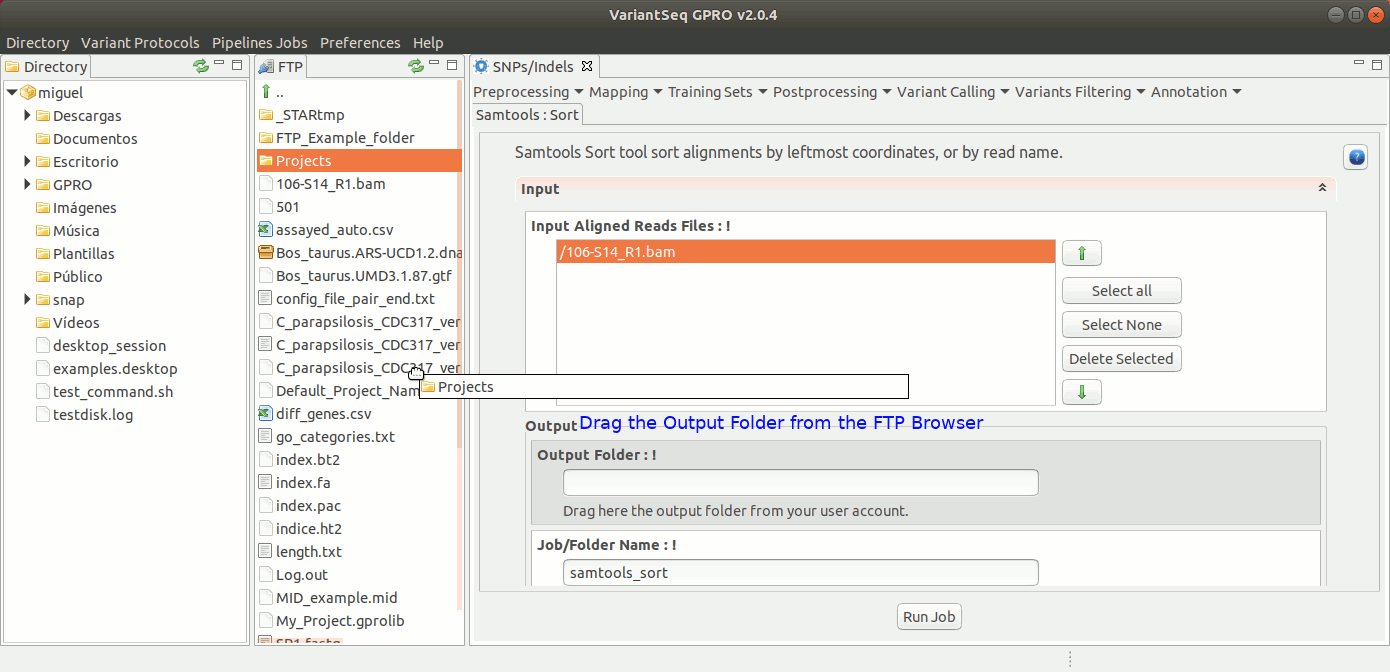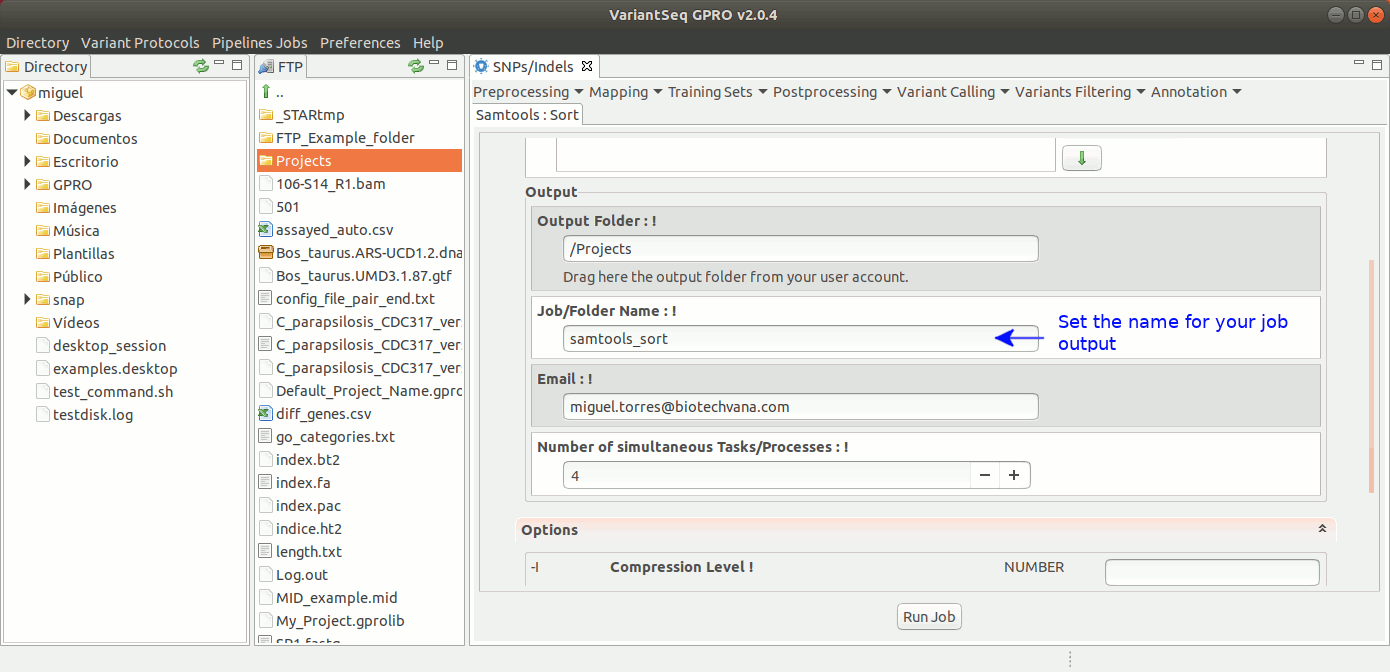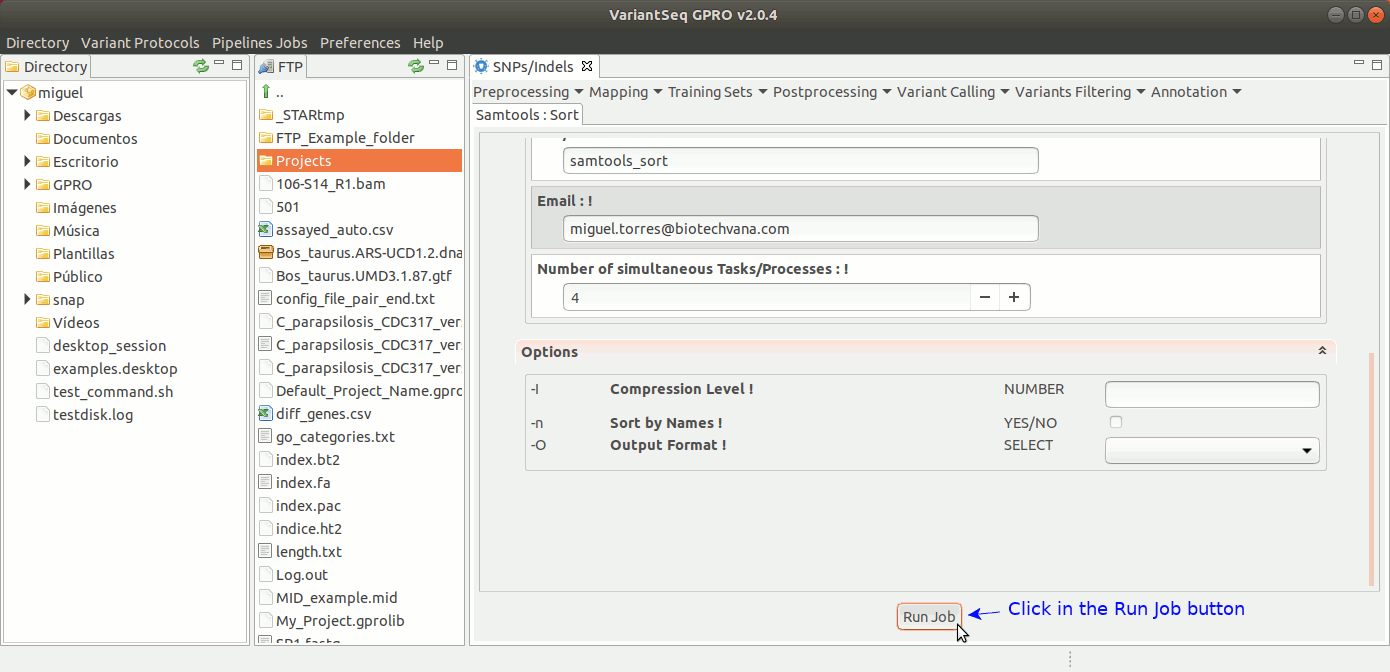
Figure 30: Using the GPRO interface for Samtools Sort.
- SAM Tools: Merge
SamTools Merge merges multiple sorted alignment files, producing a single sorted output file that contains all the input records while maintaining the sort order. For more details see the samtools manual at http://samtools.sourceforge.net
To run Samtools Merge, go to [ Postprocessing → Samtools → Merge ] and follow Fig. 31.
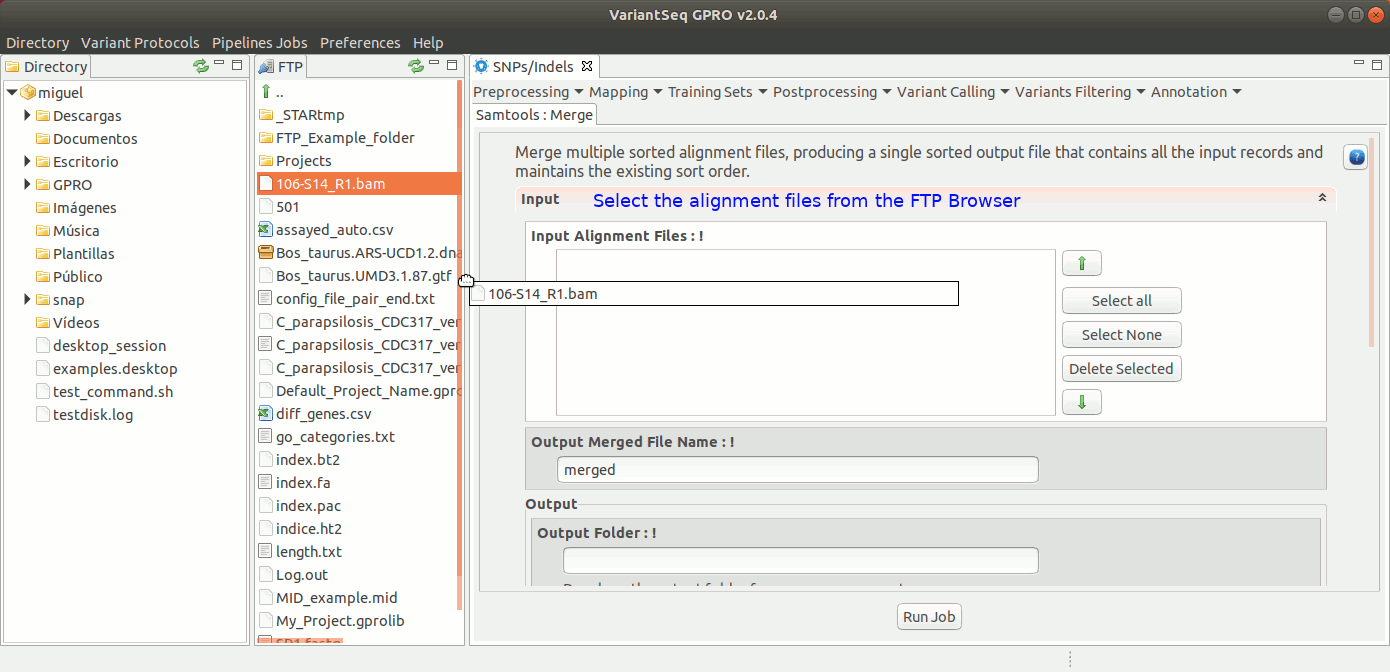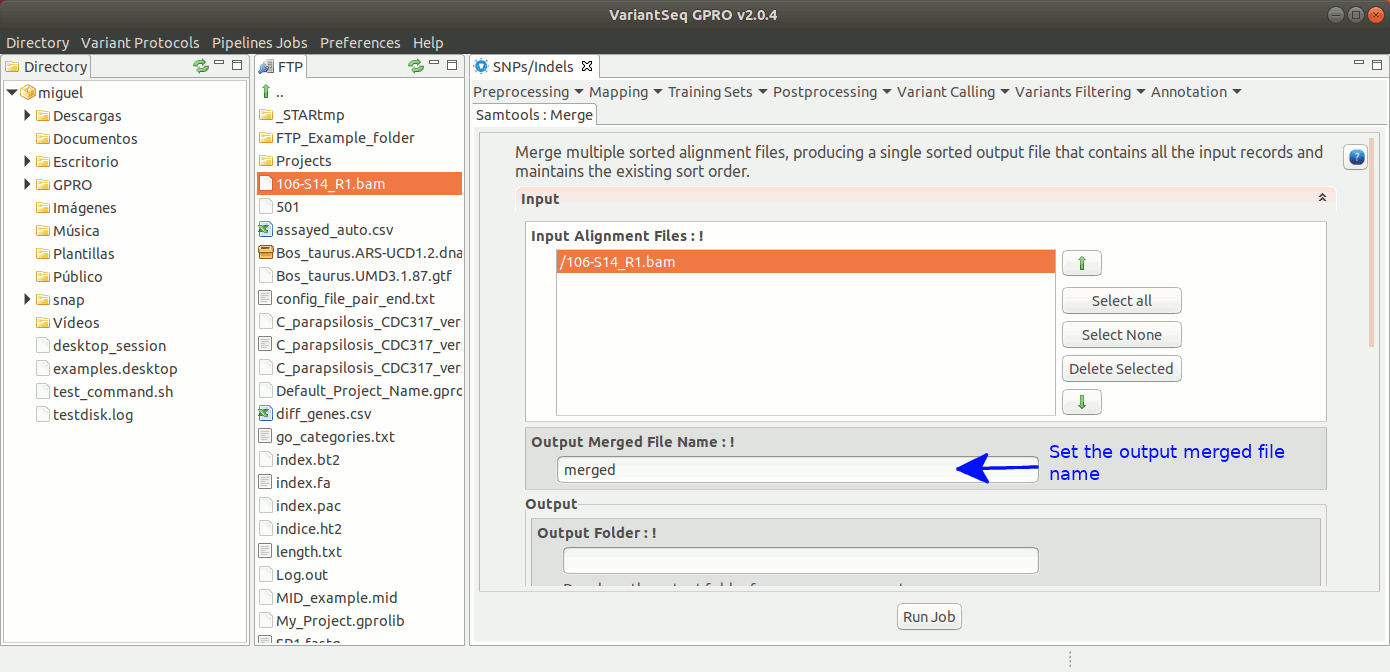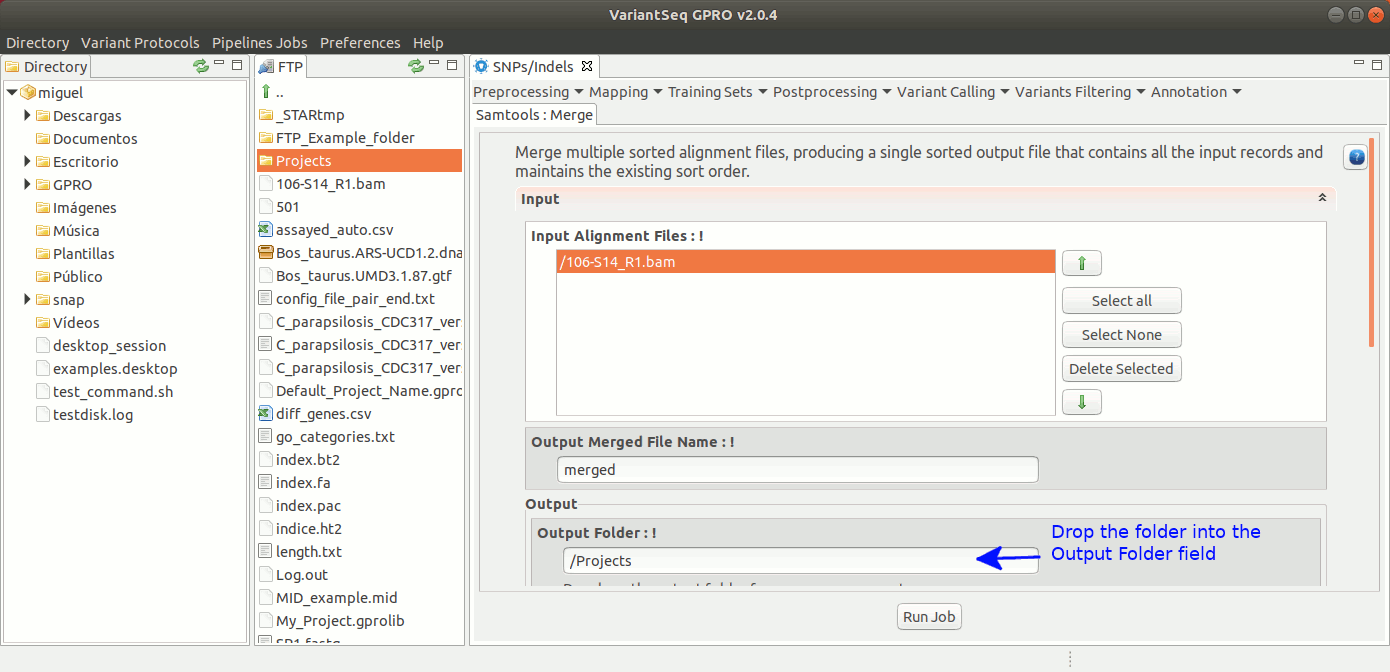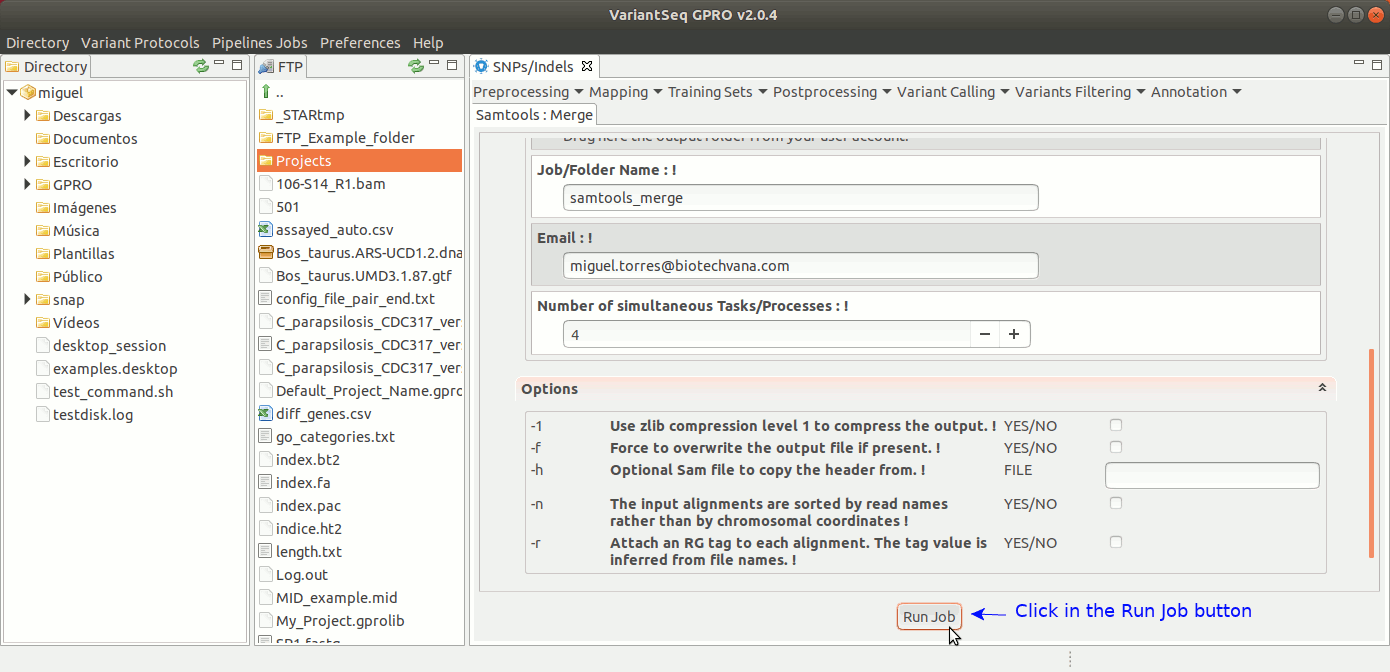
Figure 31: Using the GPRO interface for Samtools Merge.
VariantSeq provides distinct options to call SNPs and Indels based on different tools of GATK (McKenna et al. 2010; Cibulskis et al 2013; DePristo, 2011) and VarScan2 (Koboldt et al. 2012). These tools are available via the “Calling” tab in the VariantSeq menu. The menu provide distinct interfaces to manage the two set of tools provided by GATK and VarScan.
- GATK based: HaplotypeCaller
The HaplotypeCaller of GATK calls germline SNPs and indels via local re-assembly of haplotypes. For more details see the GATK Forum
To run HaplotypeCaller, go to [ Variant Calling → GATK based → HaplotypeCaller → GATK-HaplotypeCaller ] and follow Fig. 32.
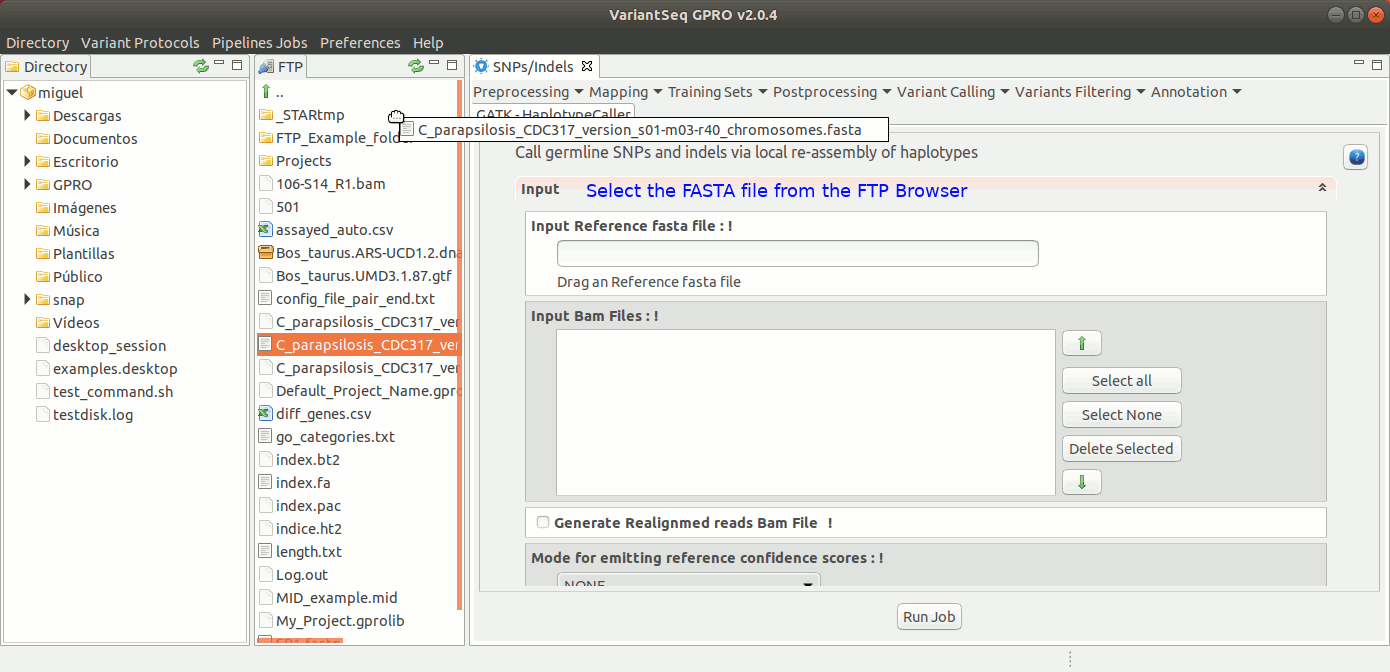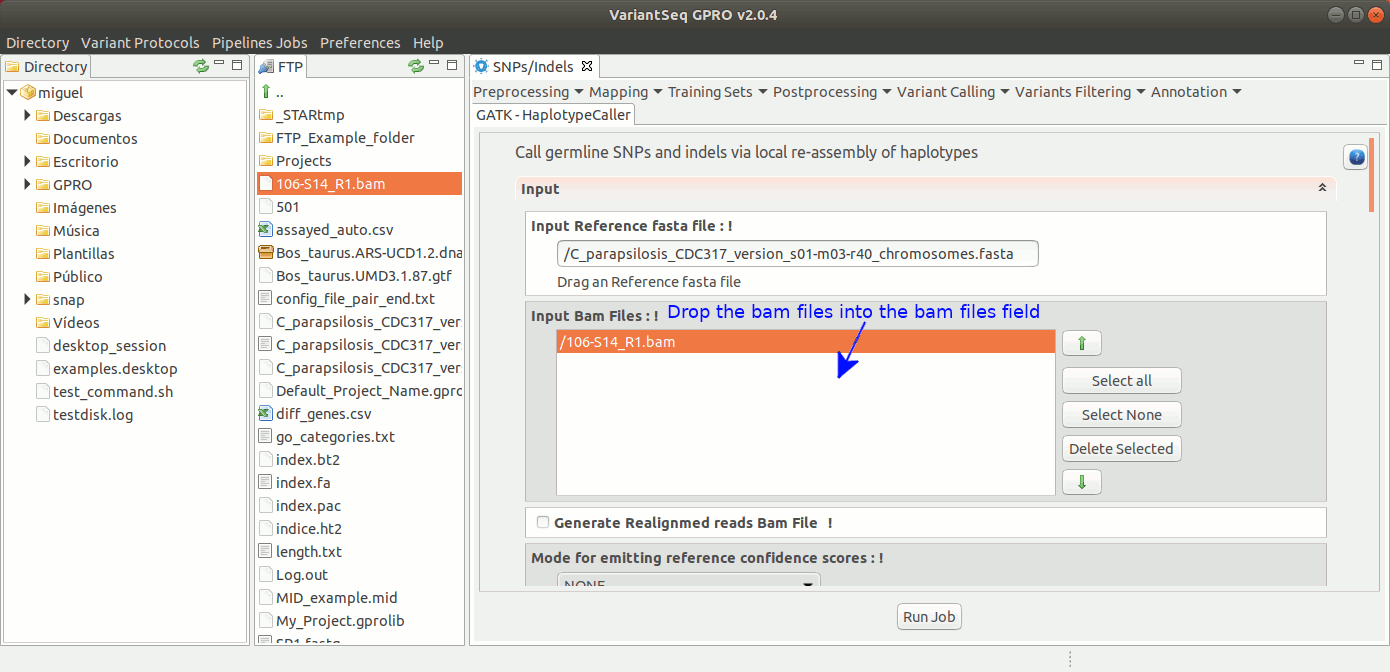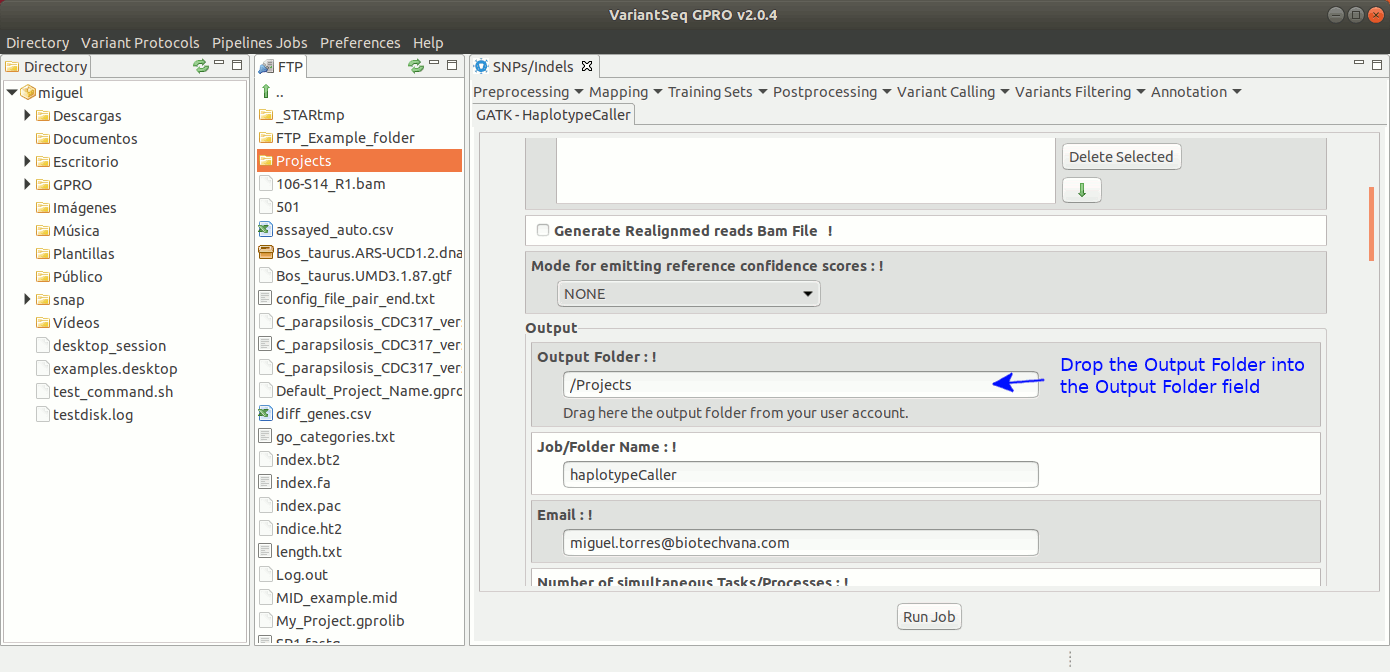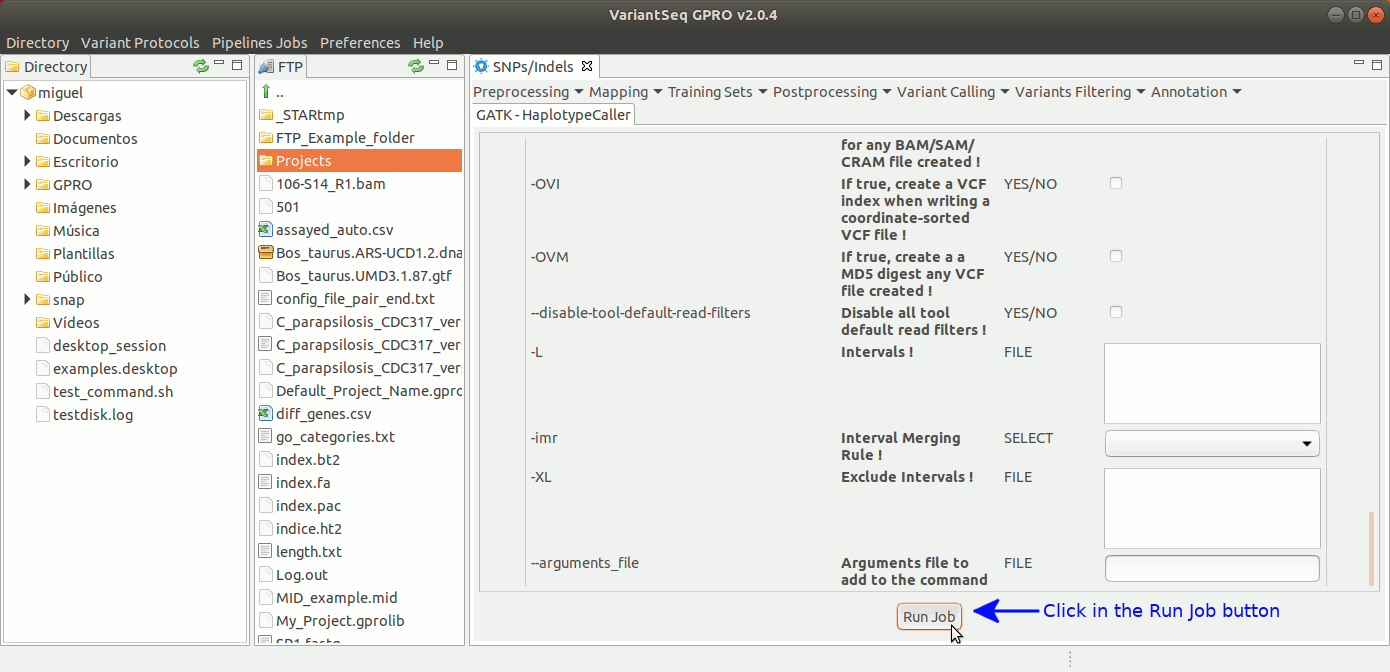
Figure 32: Using the GPRO interface for HaplotypeCaller.
- GATK based : CombineGVCFs
The HaplotypeCaller provides two other tools: “CombineGVCFs” and “GenotypeGVCFs”. CombineGVCFs merges one or more GVCF files (i.e. genomic VCF files) generated by the HaplotypeCaller into a single GVCF with appropriate annotations. This means that you can only run Combine GVCFs after running the haplotype caller or if you have the GVCF files generated by the Haplotype caller. For more details see the GATK Forum
To run CombineGVCFs, go to [ Variant Calling → GATK based → HaplotypeCaller → GATK-CombineGVCFs ] and follow Fig. 33.
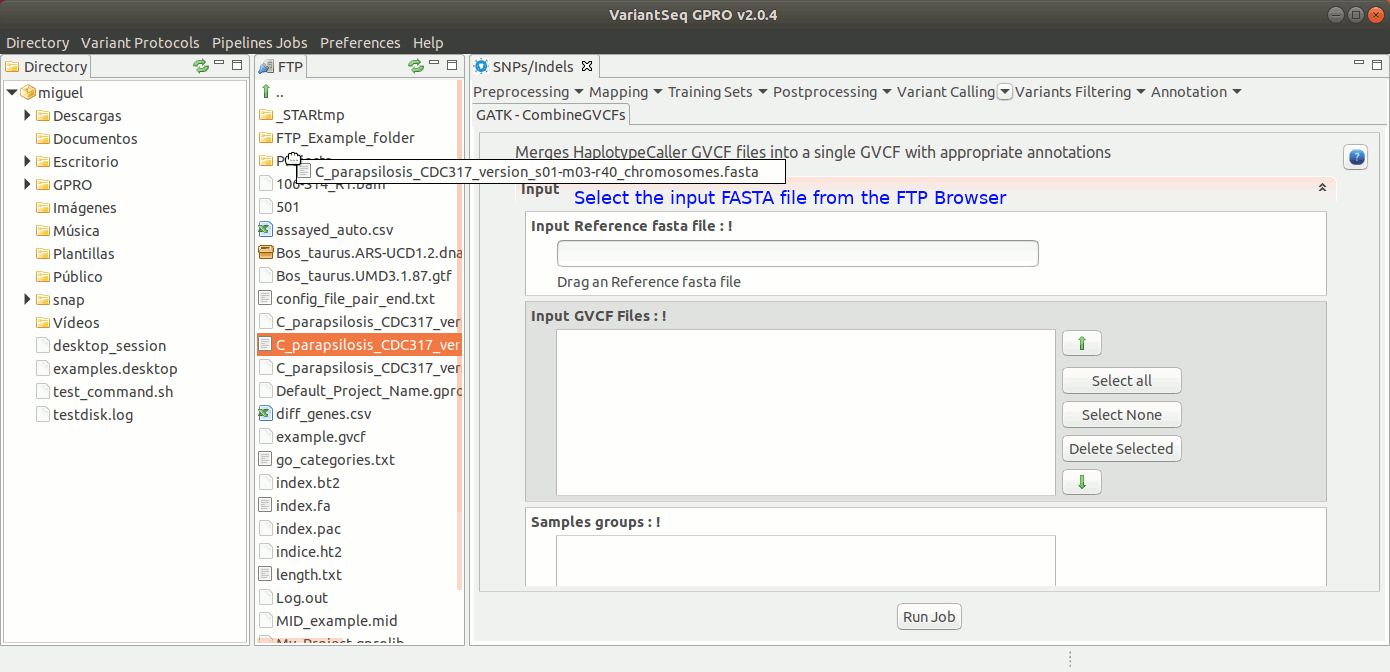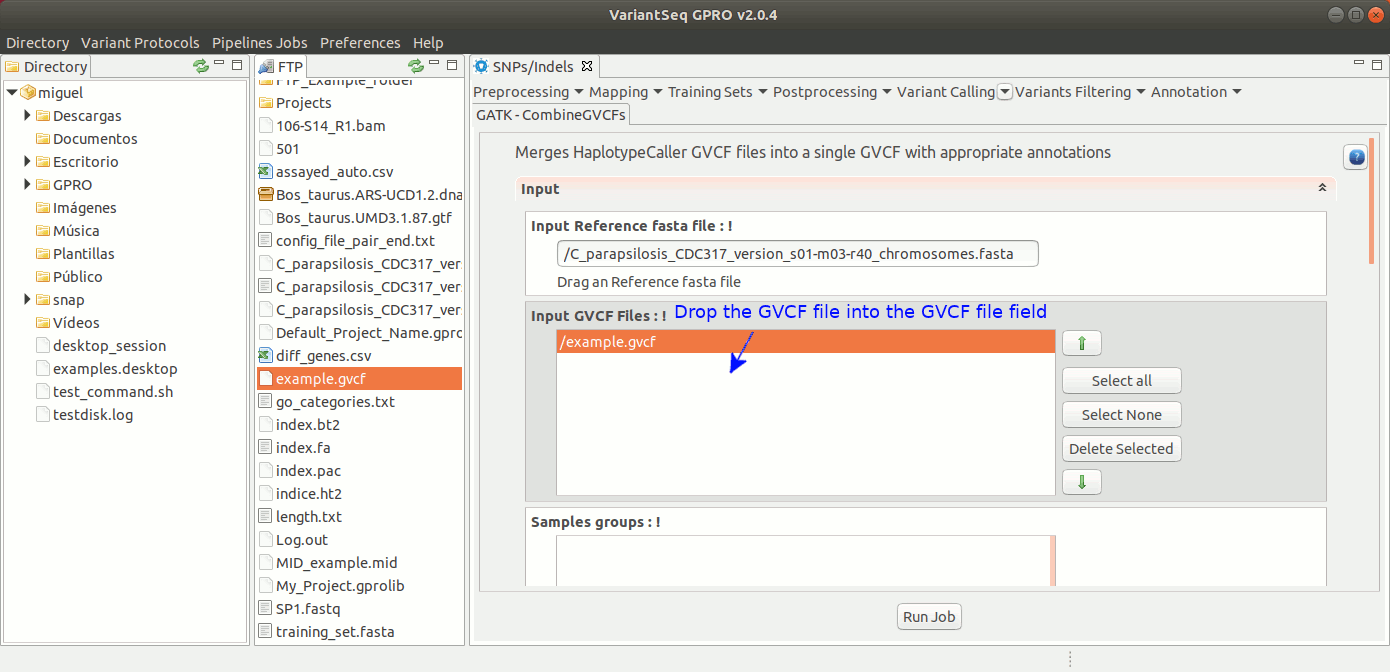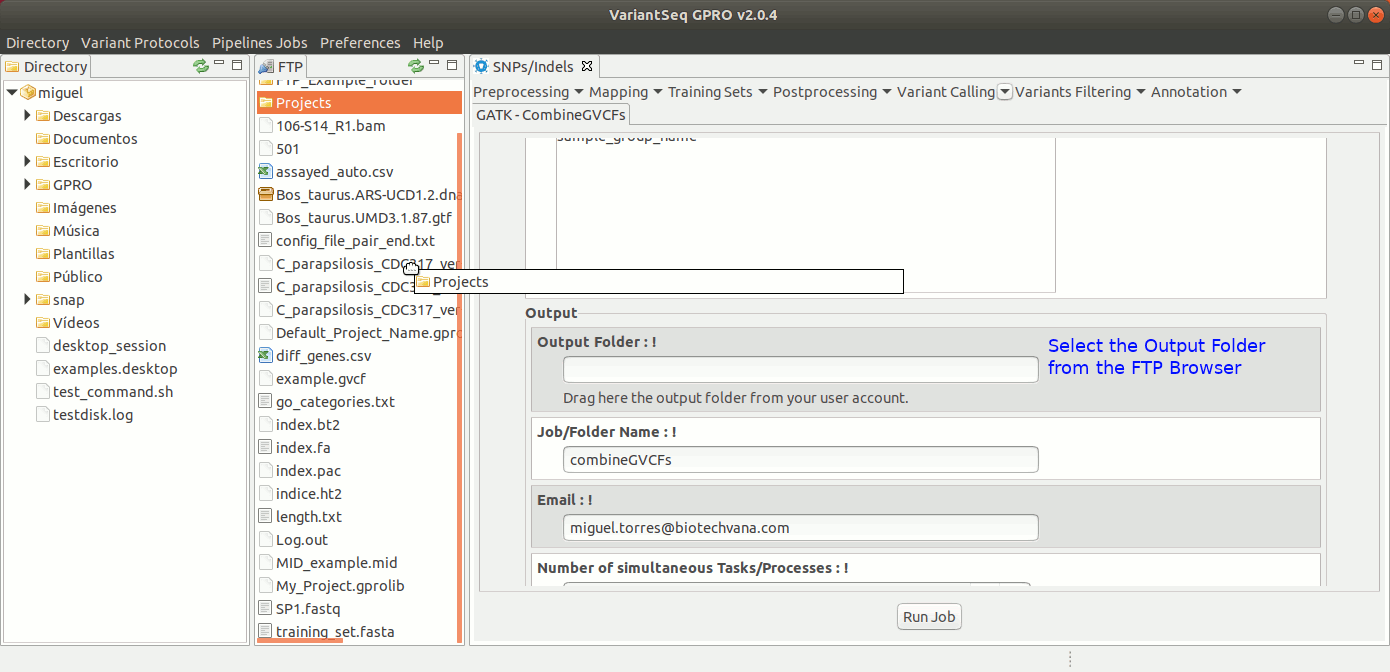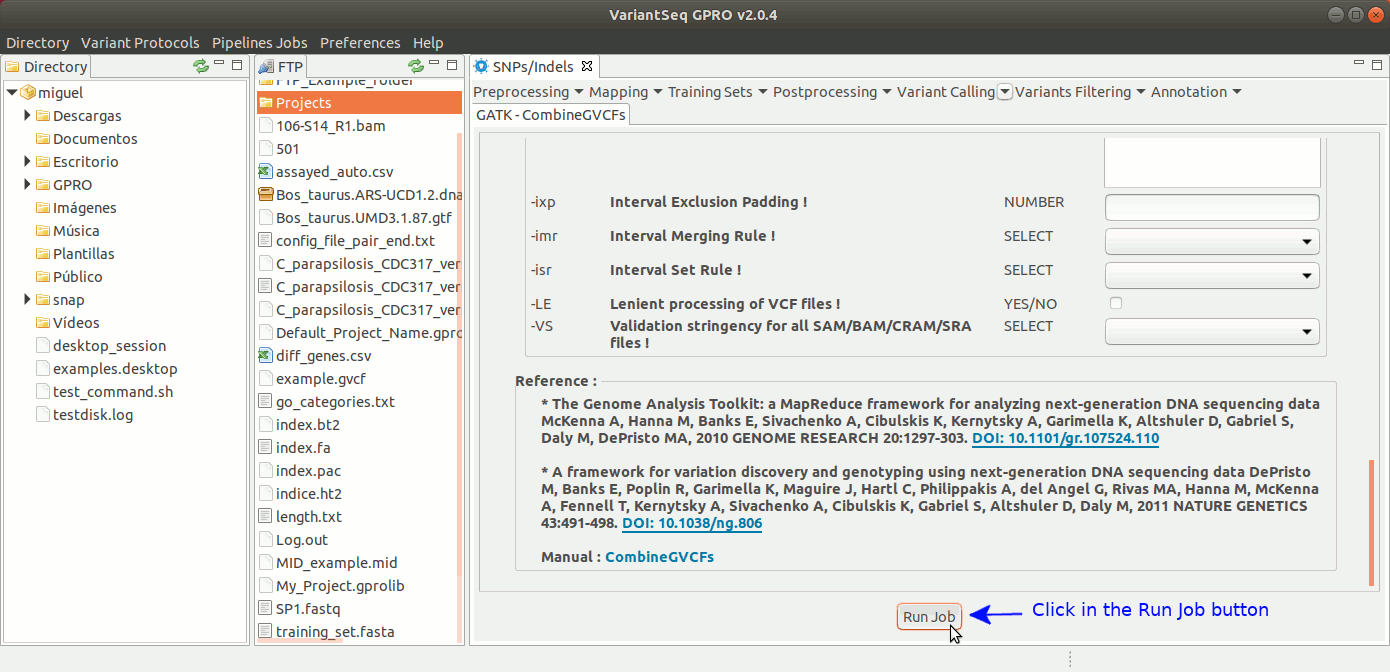
Figure 33: Using the GPRO interface for CombineGVCFs.
- GATK based : GenotypeGVCFs
GenotypeGVCFs performs joint genotyping on one or more samples pre-called with HaplotypeCaller. For more details see the GATK Forum
To run GenotypeGVCFs, go to [ Variant Calling → GATK based → HaplotypeCaller → GATK-GenotypeGVCFs ] and follow Fig. 34.
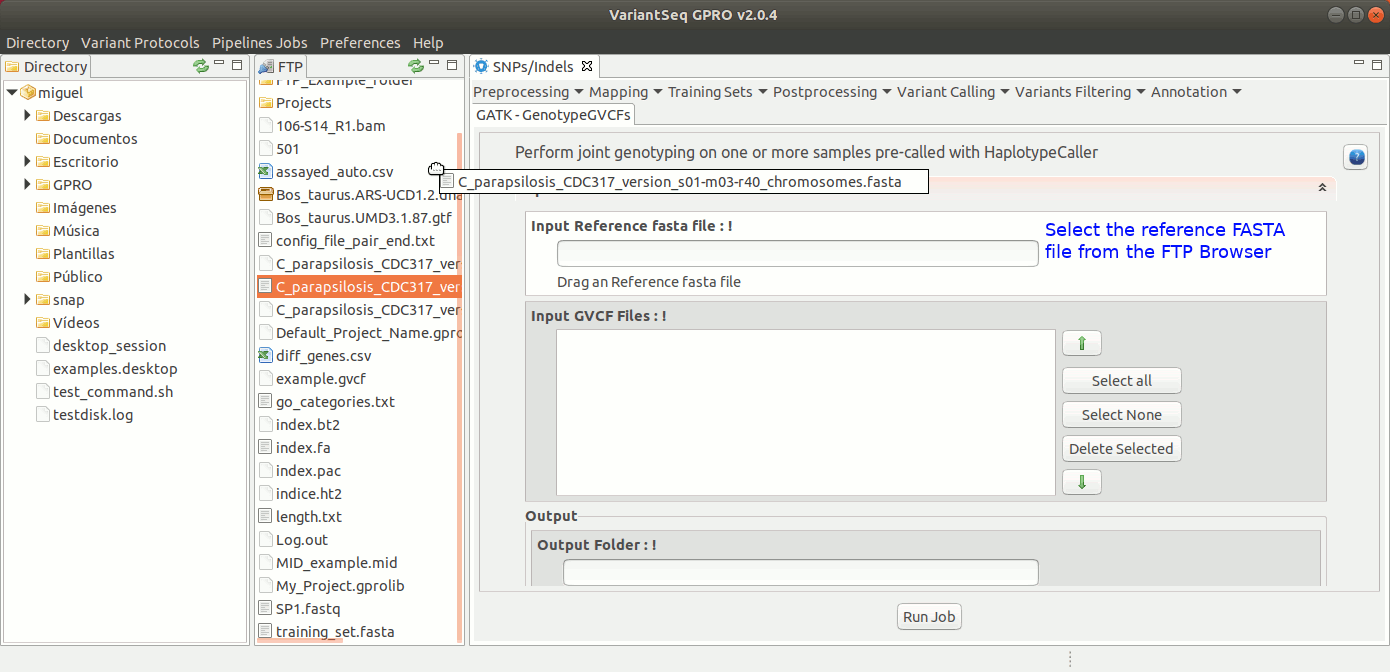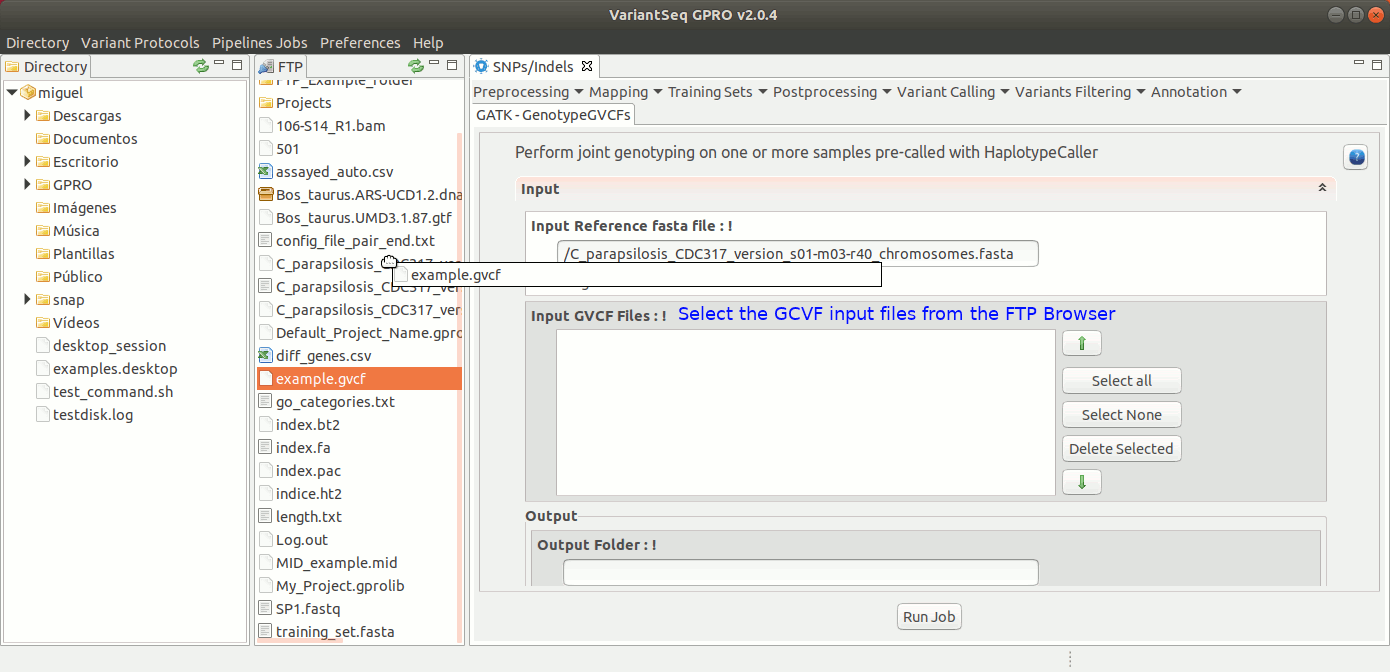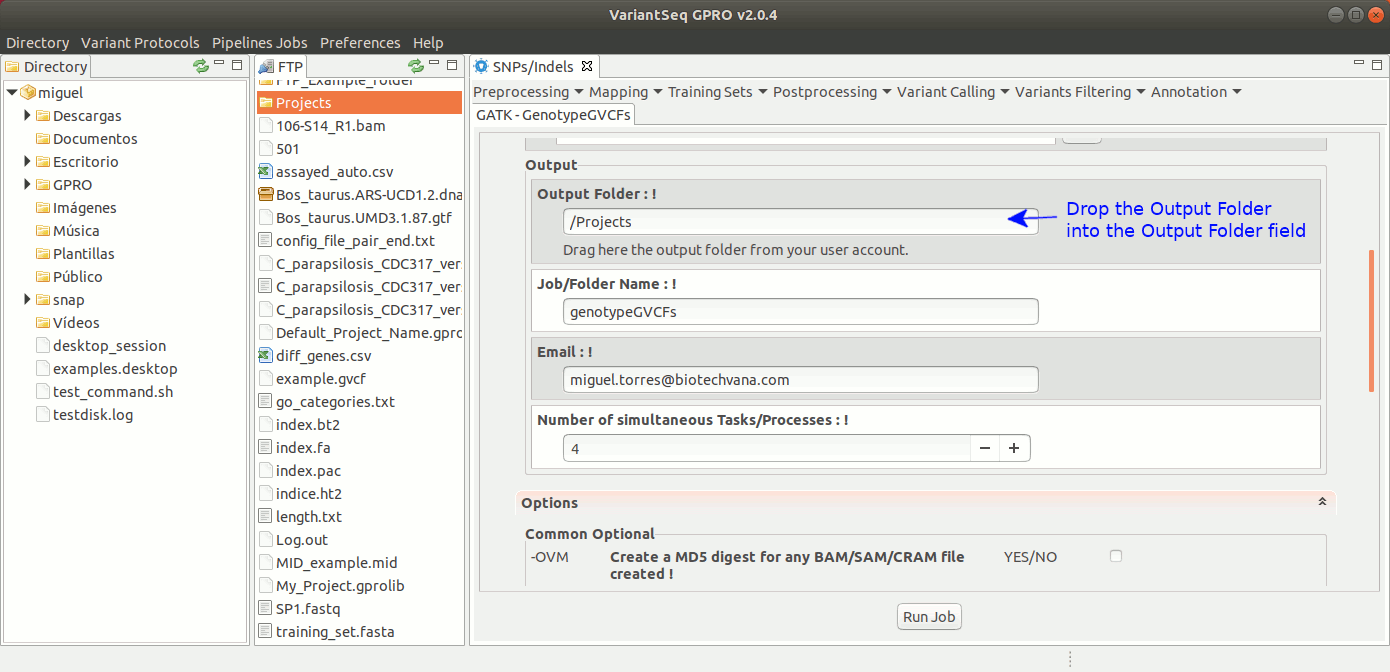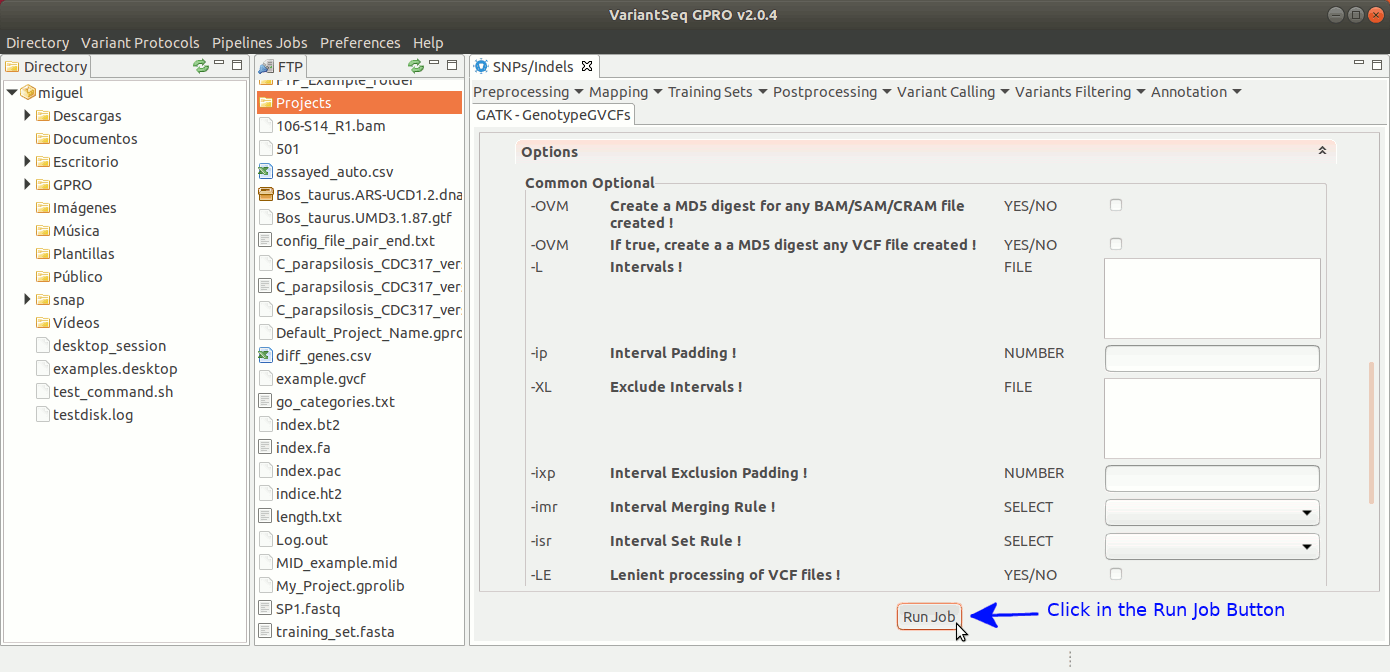
Figure 34: Using the GPRO interface for GenotypeGVCFs.
- GATK based : Mutect2
Mutect2 (McKenna et al. 2010; Cibulskis et al 2013; DePristo, 2011) calls somatic or cancer via local assembly of haplotypes allowing to manage tumor-normal pairs (pairs of samples tumoral sample of a patient and another sample from a tissue of the same patient, not affected by the tumor) and PONs (panel of normals). For more details see the GATK forum at: GATK forum
To run Mutec2, go to [ Variant Calling → GATK based → Mutect2 ] and follow Fig. 35.
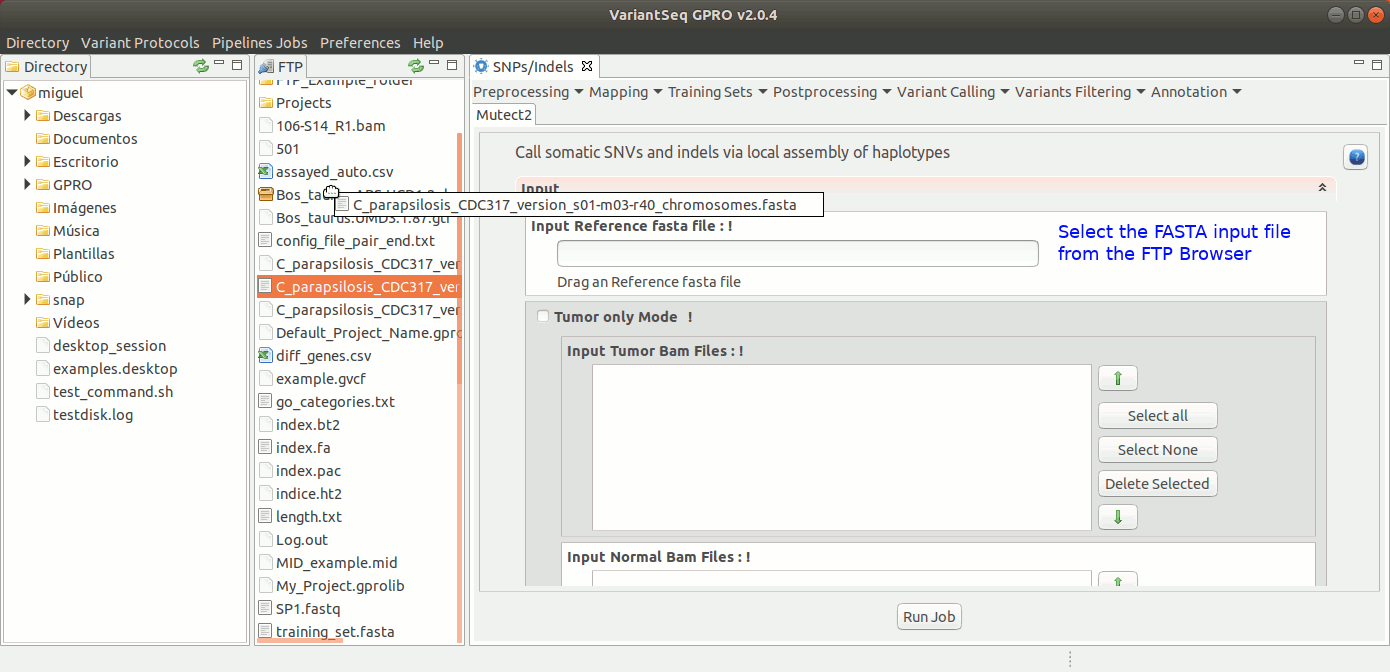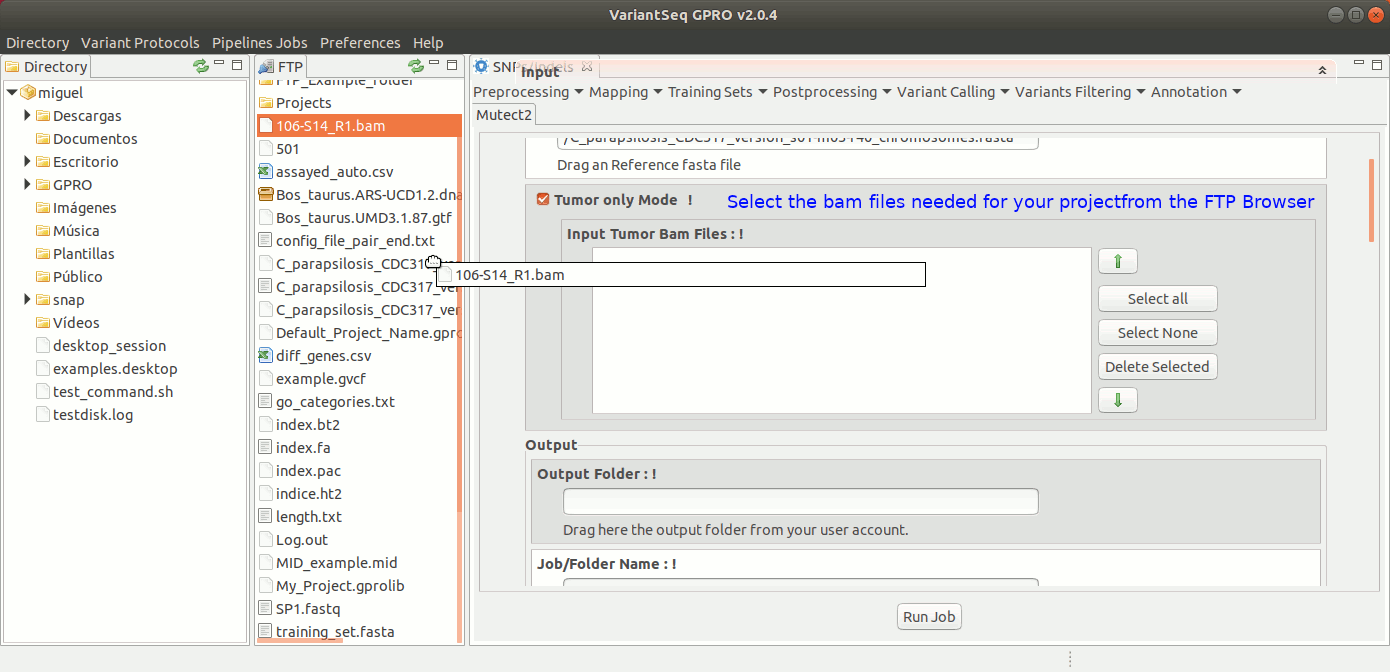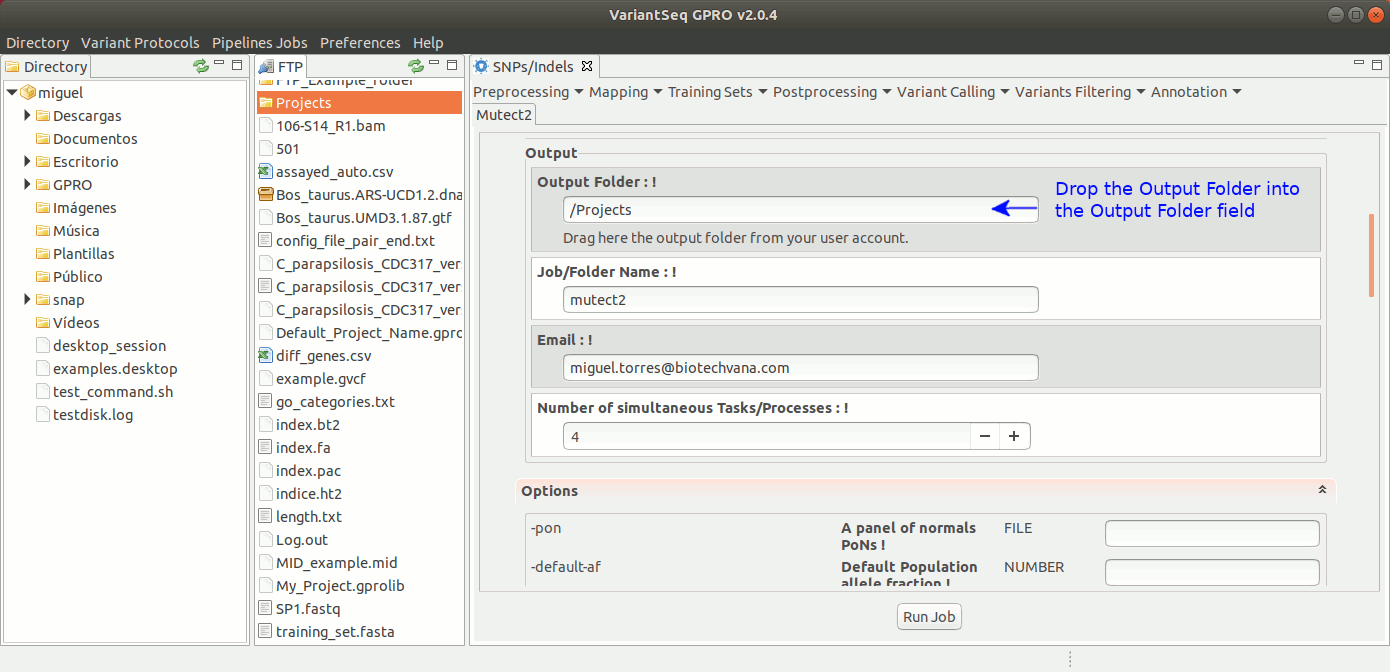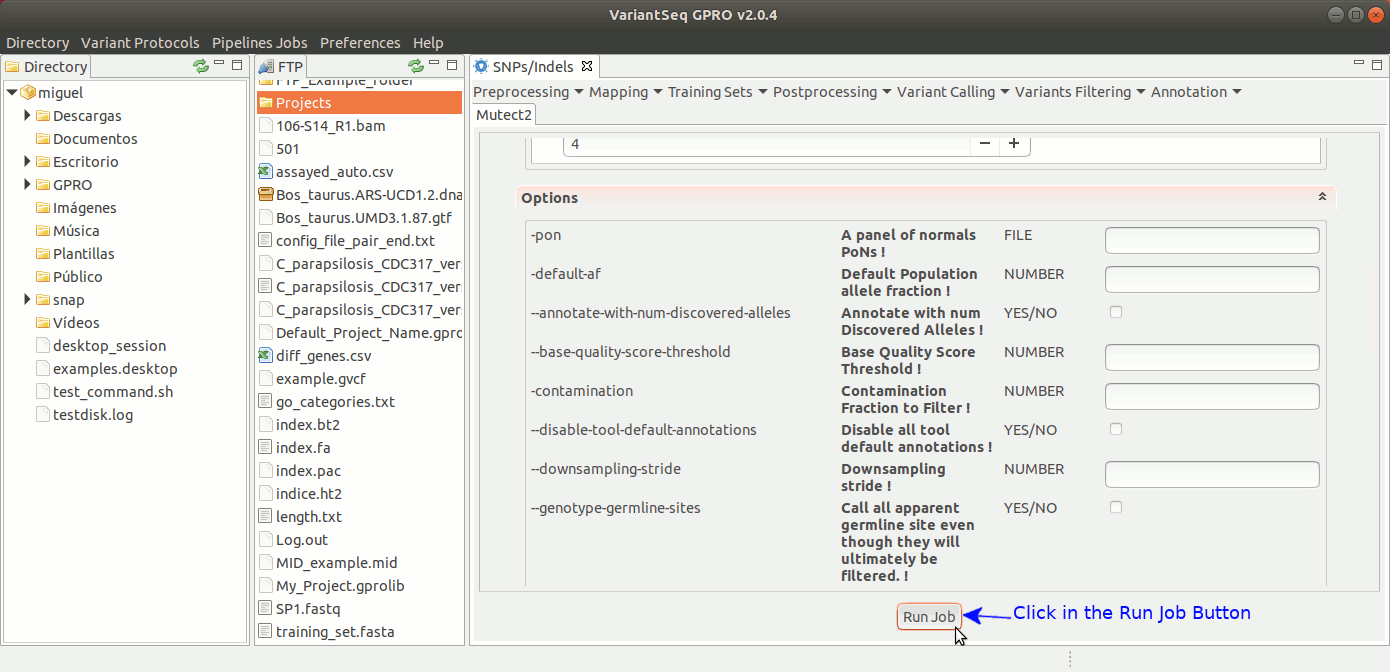
Figure 35: Using the GPRO interface for Mutect2.
VariantSeq permits calling germinal and somatic variants from one or multiple sample files (multisample calling) including those combining VarScan2 and SamTools. For more information, see the Varscan2 manual at http://varscan.sourceforge.net/using-varscan.html.php.
- VarScan2 based : Germline variants
This protocol uses the VarScan2 germline variants tool and SamTools to enable multisample calling of germinal SNPs and Indels. Basically, SamTools generates a pileup file that is subsequently parsed by VarScan2 to compute the number of bases supporting each observed allele. For more details see the VarScan2 manual at : https://dkoboldt.github.io/varscan/germline-calling.html.
To run VarScan2 and SamTools for germline variant calling, go to [ Variant Calling → VarScan2 basd → Germline variants ] and follow Fig. 36.
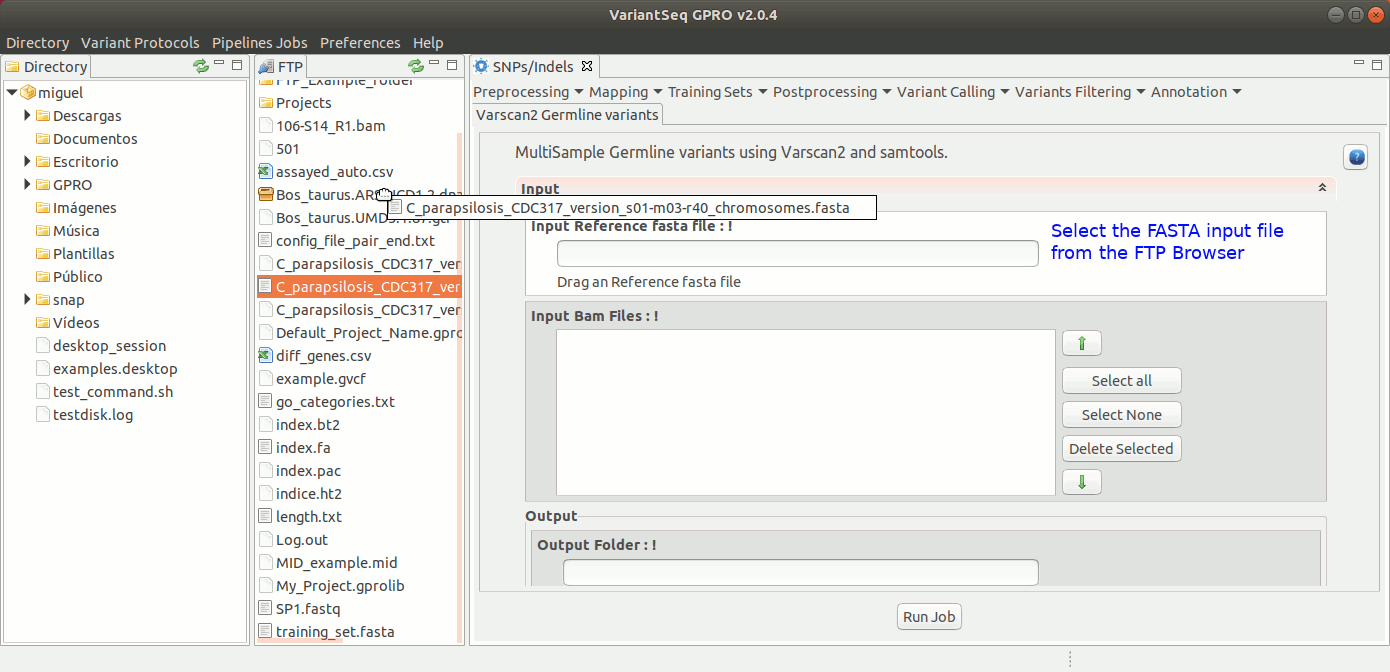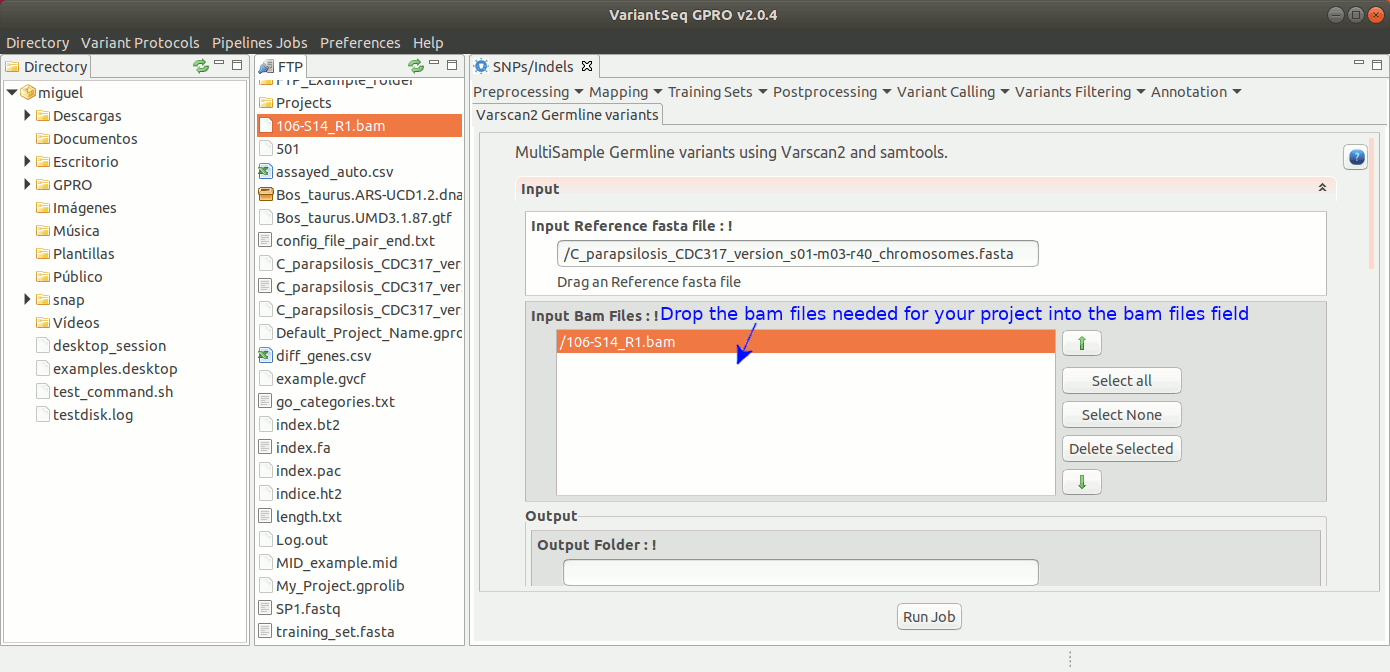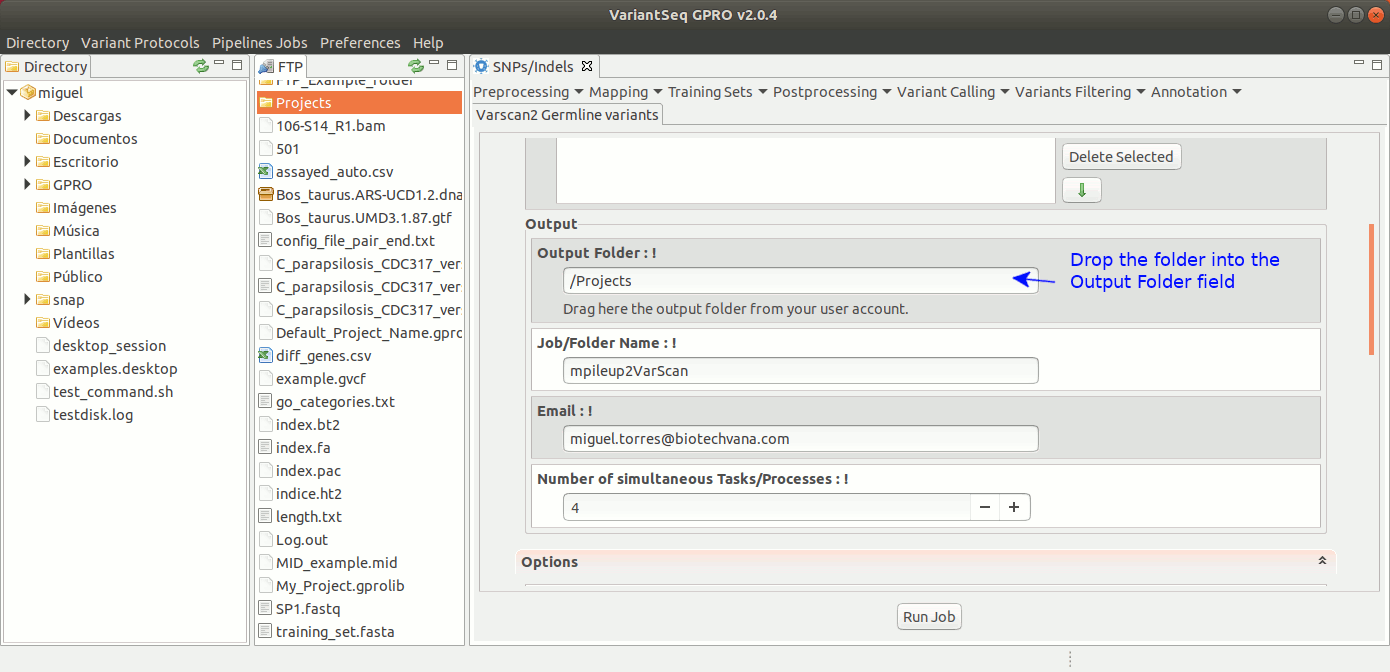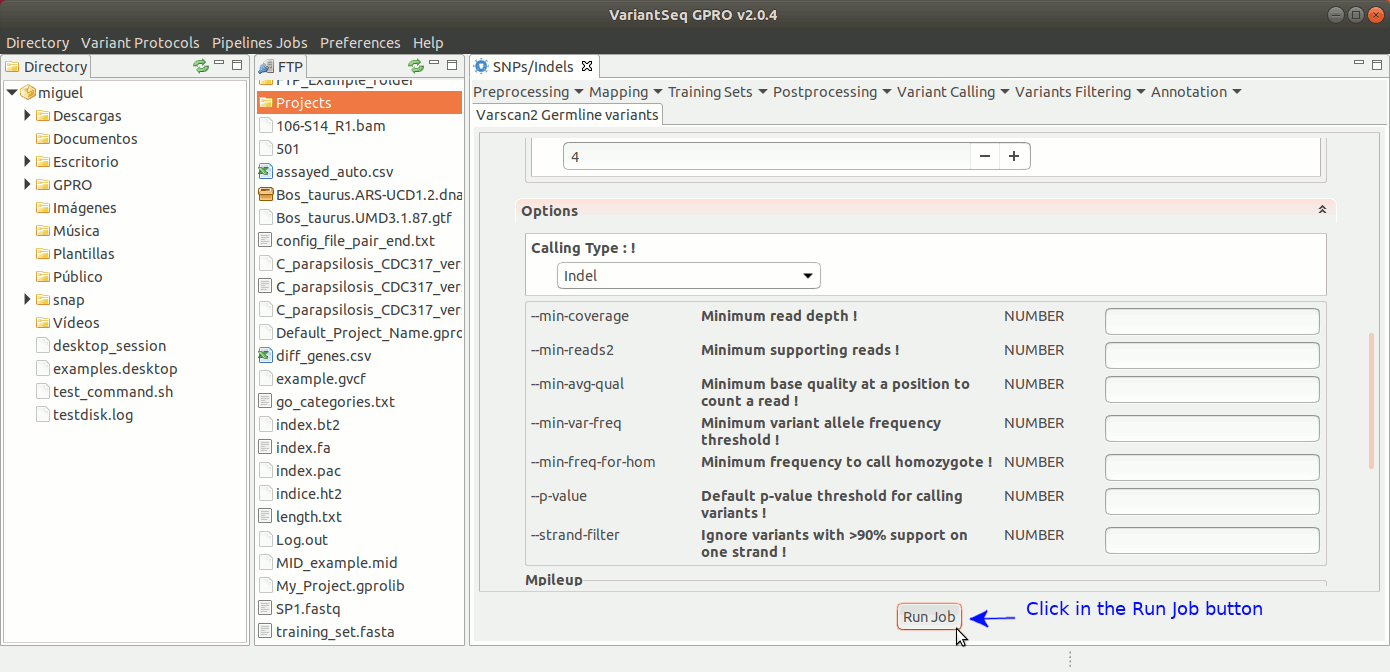
Figure 36: Using the GPRO interface for VarScan2 germline variants plus SamTools.
- VarScan2 based: Somatic variants
This protocol uses VarScan2 somatic variants and Samtools to perform multisample calling of somatic SNPs and Indels using normal-tumour pairs (pairs of samples tumoral sample of a patient and another sample from a tissue of the same patient, not affected by the tumor). SamTools generates a pileup file that is used by VarScan2 to call somatic variants (SNPs and indels) using a heuristic method and a statistical test based on the number of aligned reads supporting each allele. For more details see the VarScan2 manual at : http://varscan.sourceforge.net/somatic-calling.html.
To run VarScan2 and SamTools for somatic variant calling, go to [Variant Calling → VarScan2 based → somatic variants ] and follow Fig. 37.
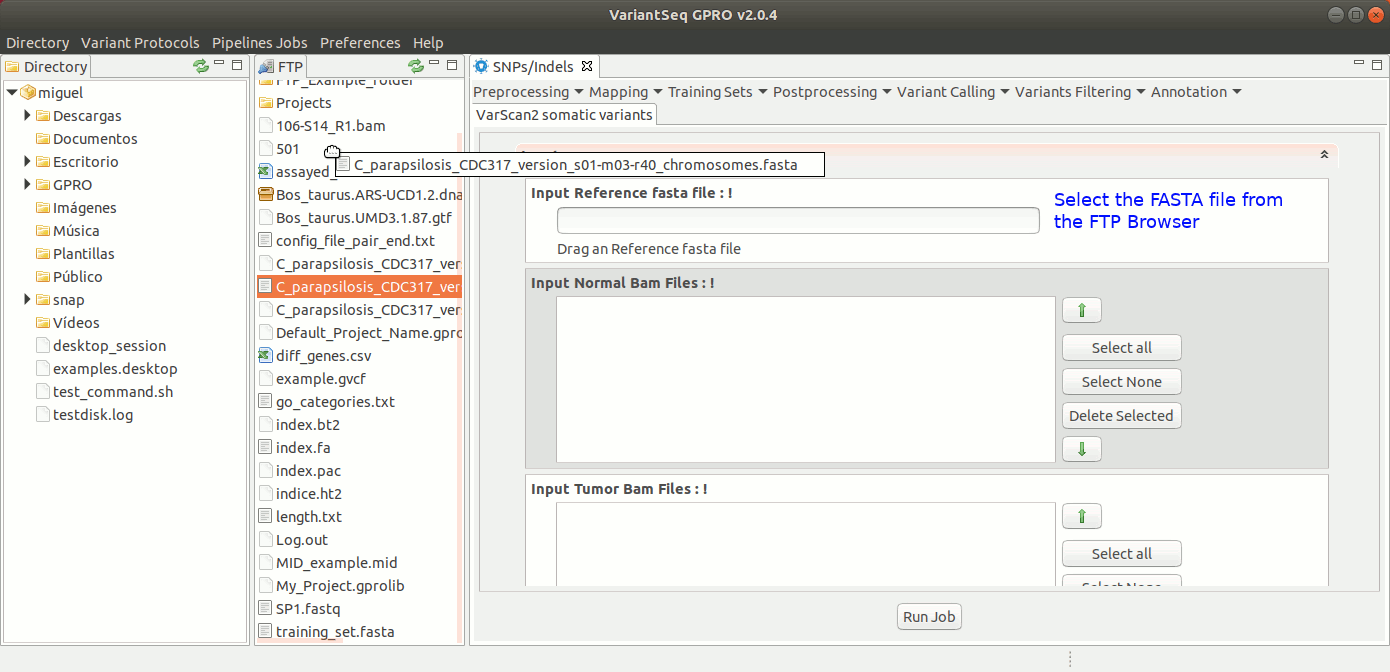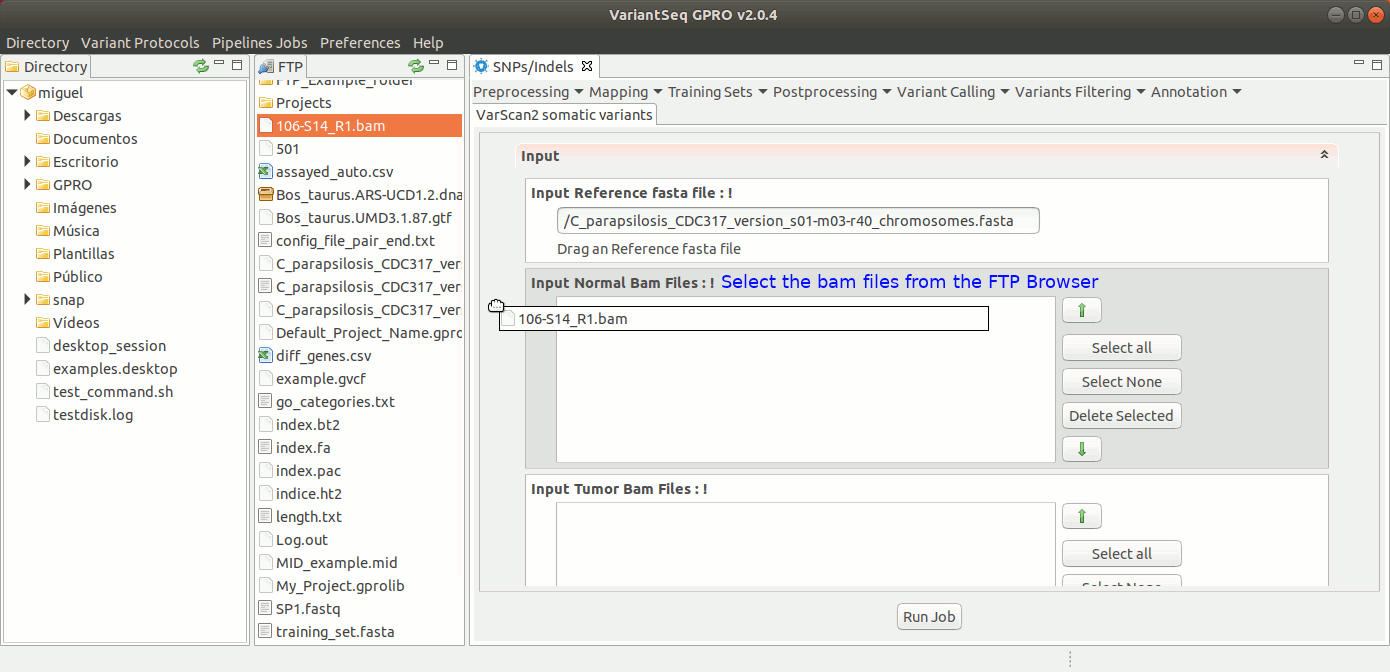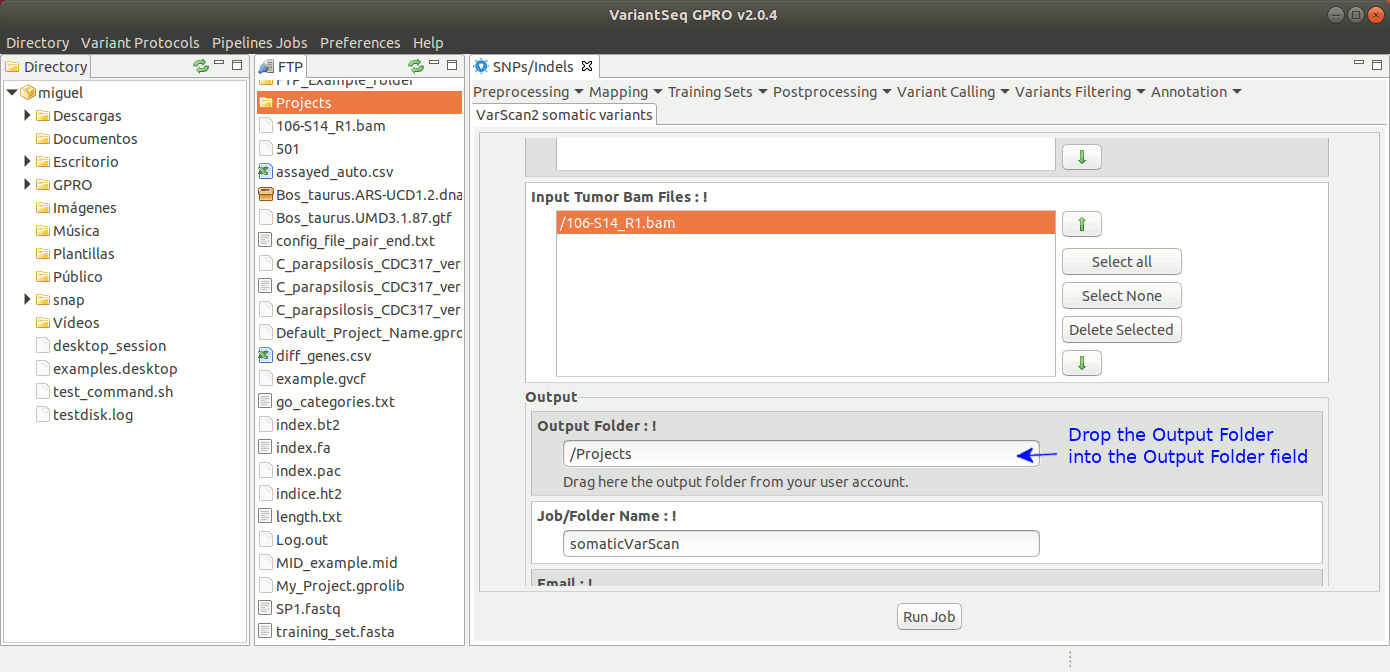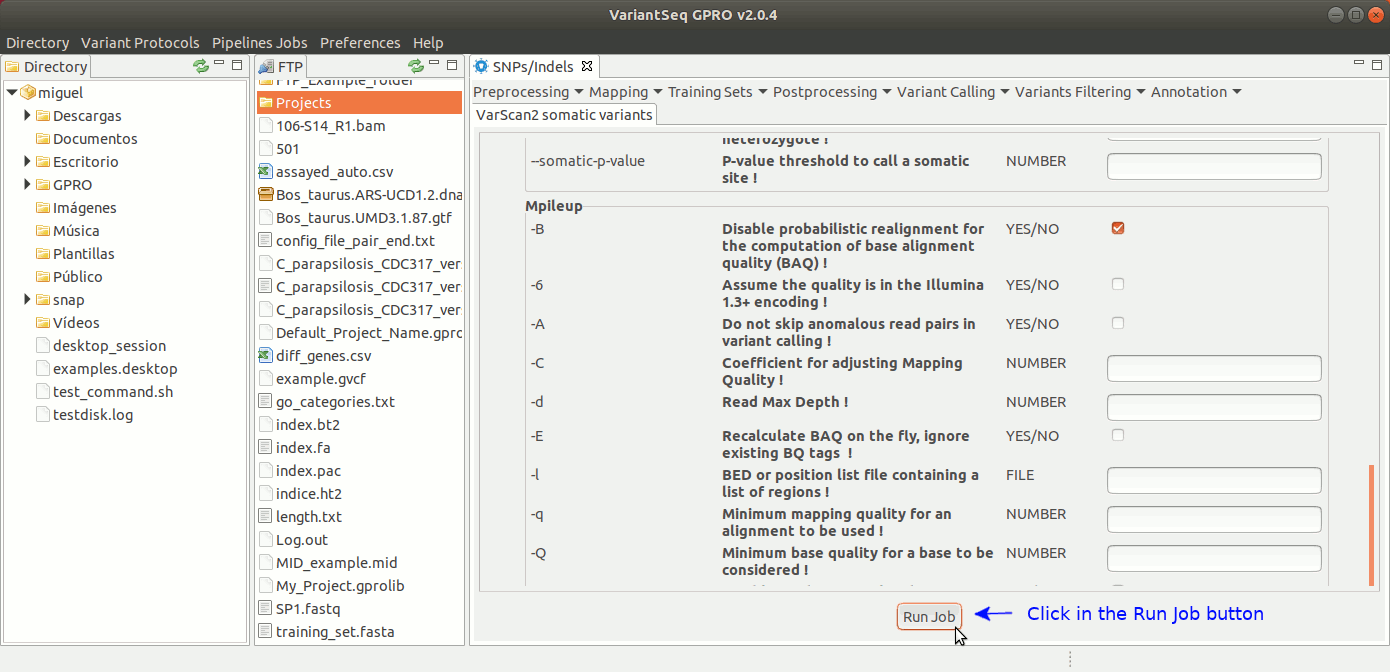
Figure 37: Using the GPRO interface for VarScan2 somatic variants plus SamTools
- VarScan2 based: Trio calling
This tool enables VarScan2 Trio and SamTools to analyze family pedigrees among three samples groups (father, mother and child) with the objective of identifying transmitted alleles and/or de novo mutations that may confer susceptibility to disease. Trio leverages the family relationship to improve variant calling accuracy, identify apparent Mendelian Inheritance Errors (MIEs), and detect de novo mutations with high confidence. For more details see the VarScan2 manual at: http://varscan.sourceforge.net/trio-calling-de-novo-mutations.html
To run samtools and VarScan2 Trio for pedigree calling, go to [ Variant Calling → VarScan2 based → Trio calling ] and follow Fig. 38.
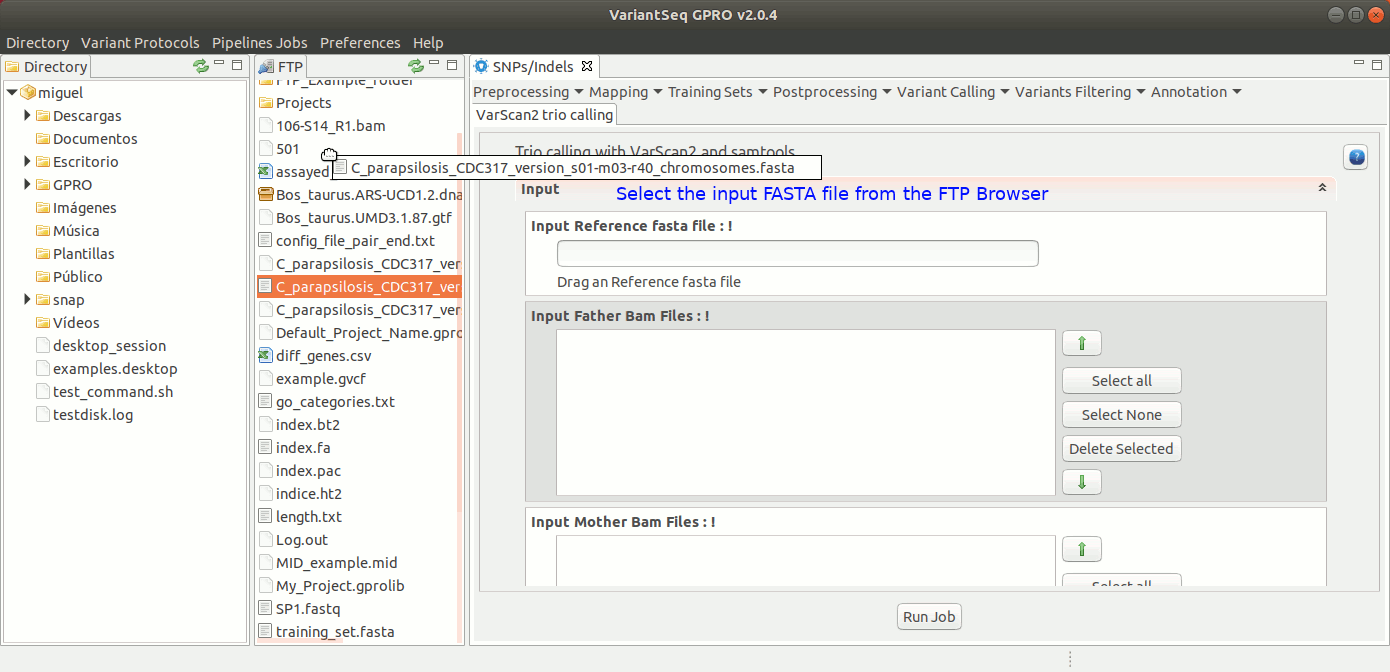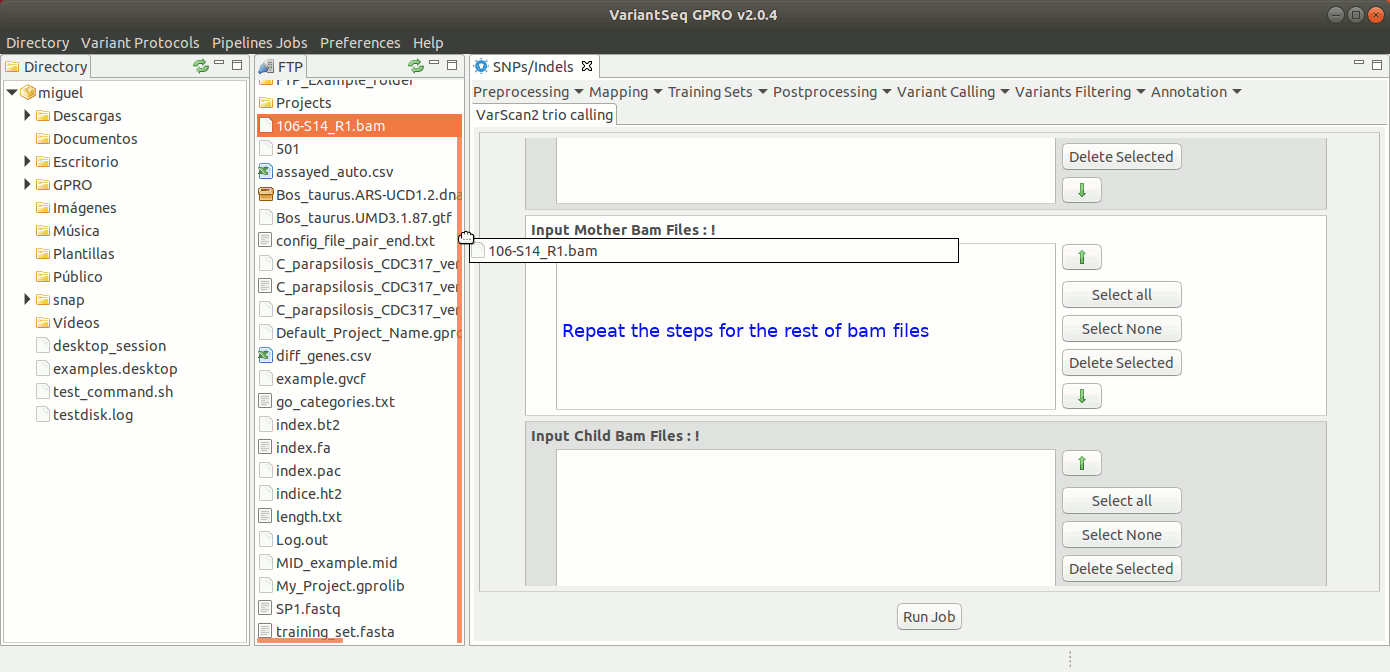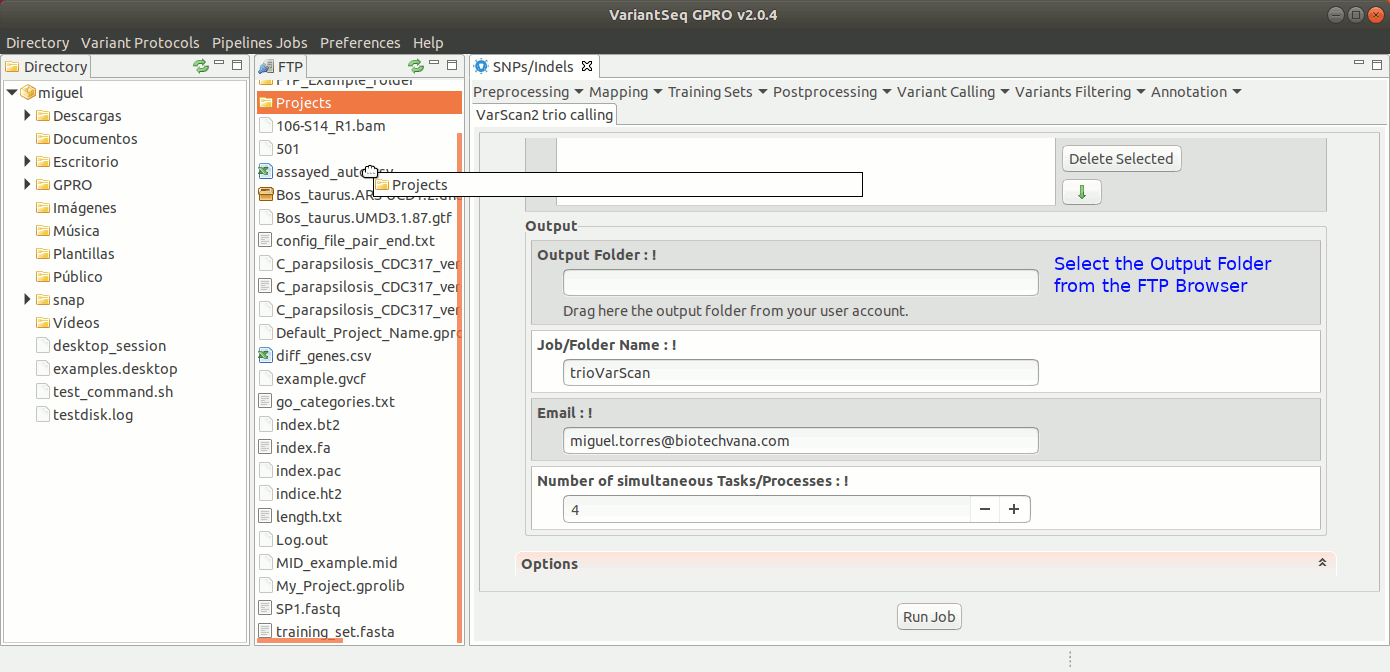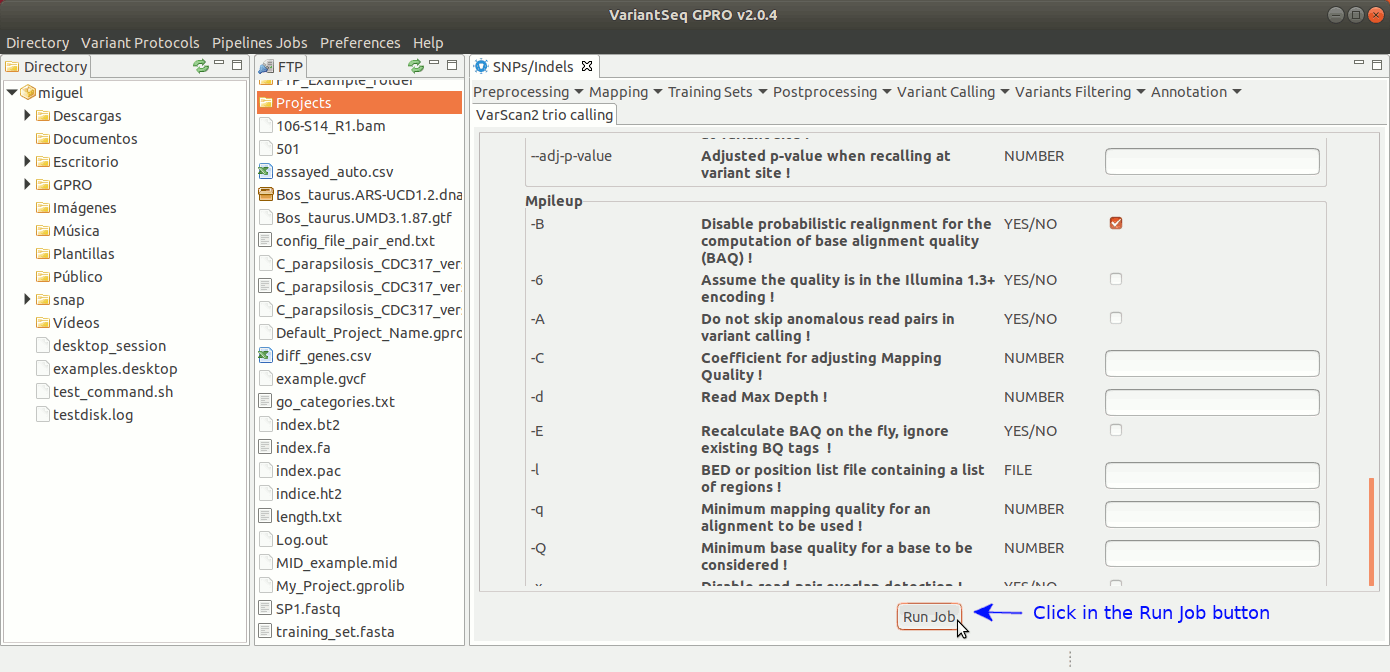
Figure 38: Using the GPRO interface for VarScan2 for Trio plus SamTools.
VariantSeq implements tools based on GATK that are required for filtering and calibrating SNPs and Indels in VCF files after the calling of variants. The referred tools are “Variant Quality Score Recalibration”, “Variant Filtration”, “Cross-Sample Contamination” and “FilterMutectCalls”.
- Variant Filtering : Variant Quality Score Recalibration
Variant Quality Score Recalibration (VQSR) is a pipeline based on the VariantRecalibrator GATK command to build a recalibration model to score variant quality for filtering purposes. For more details see: this link and this link at teh GATK forum
To run VQSR, go to [ Variant Filtering → Variant Quality Score Recalibration ] and follow Fig. 39
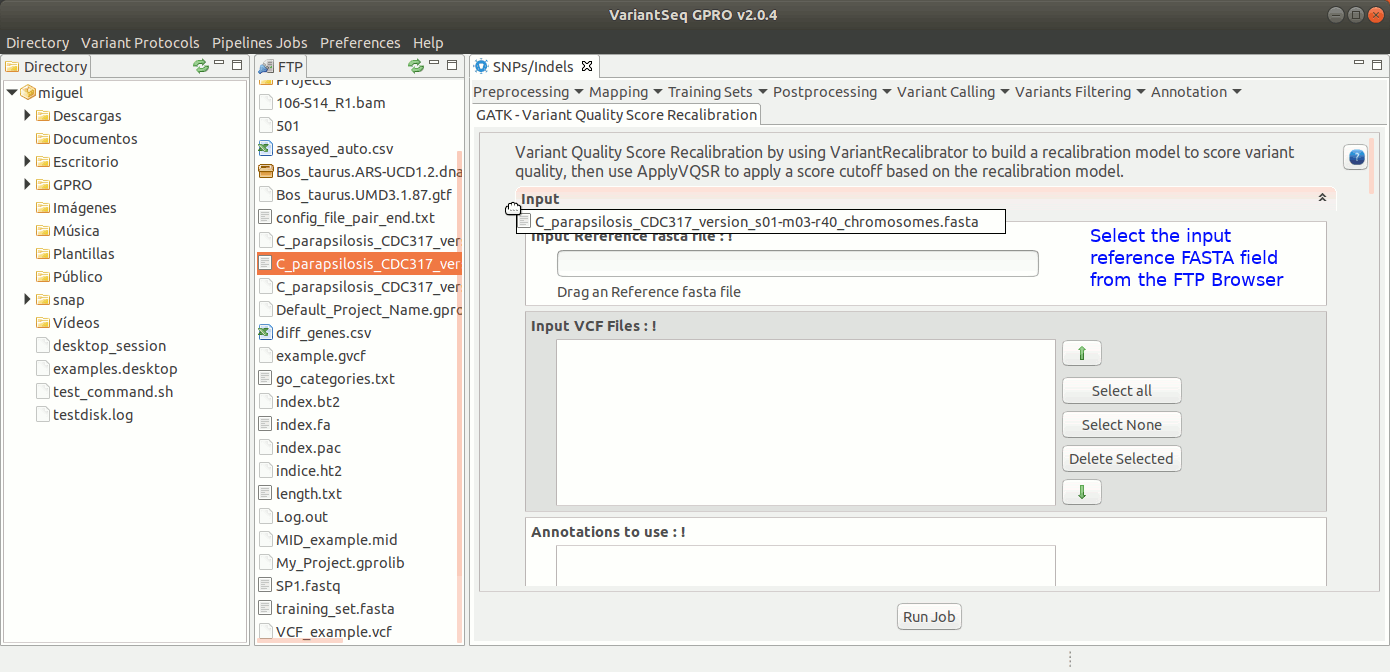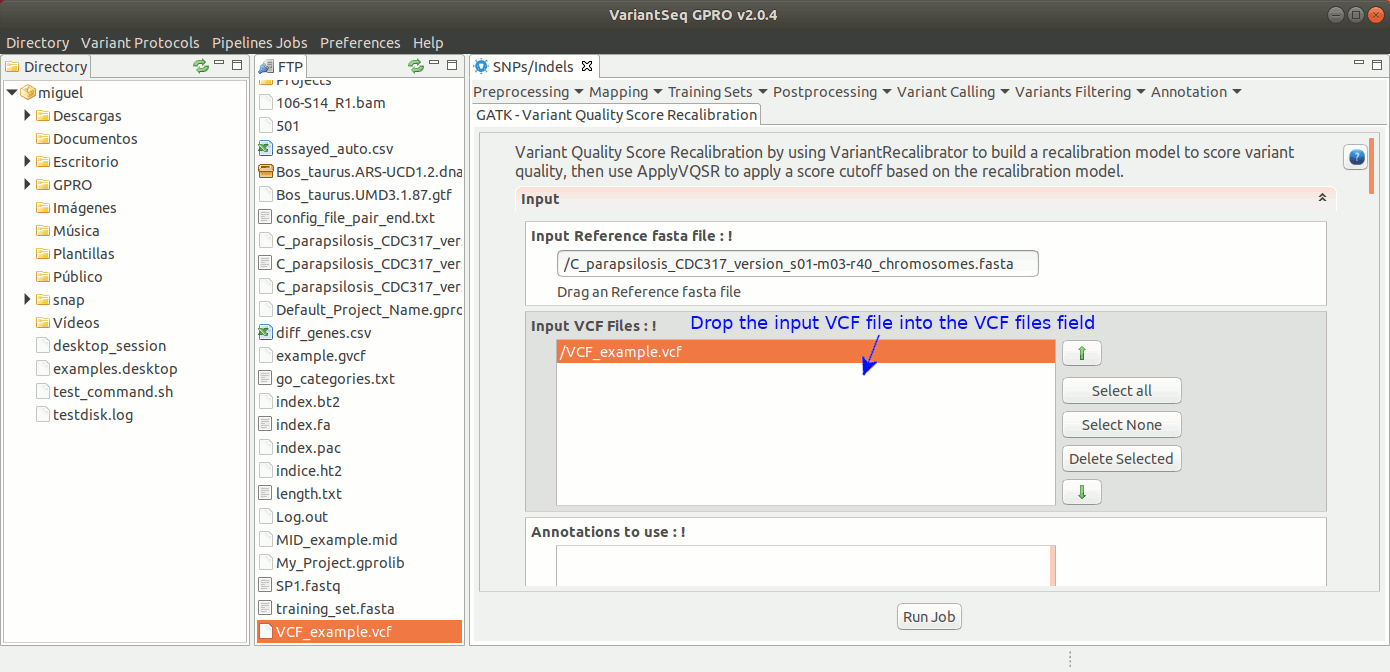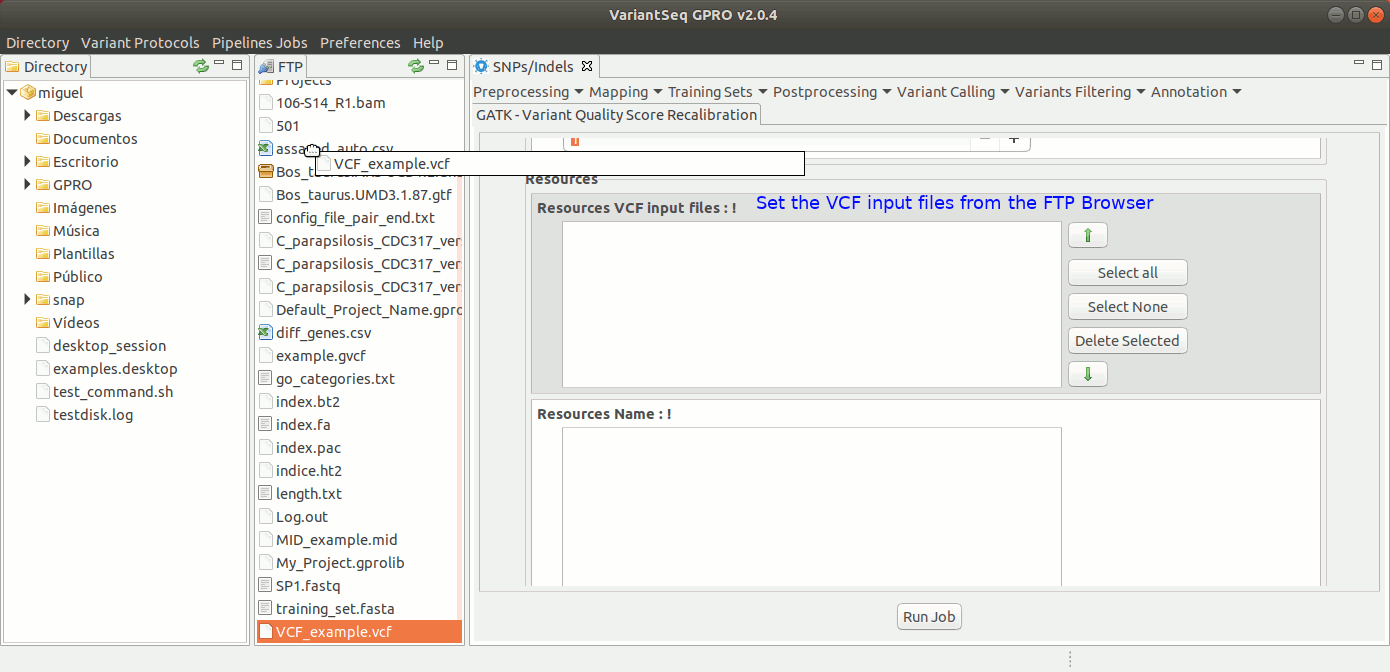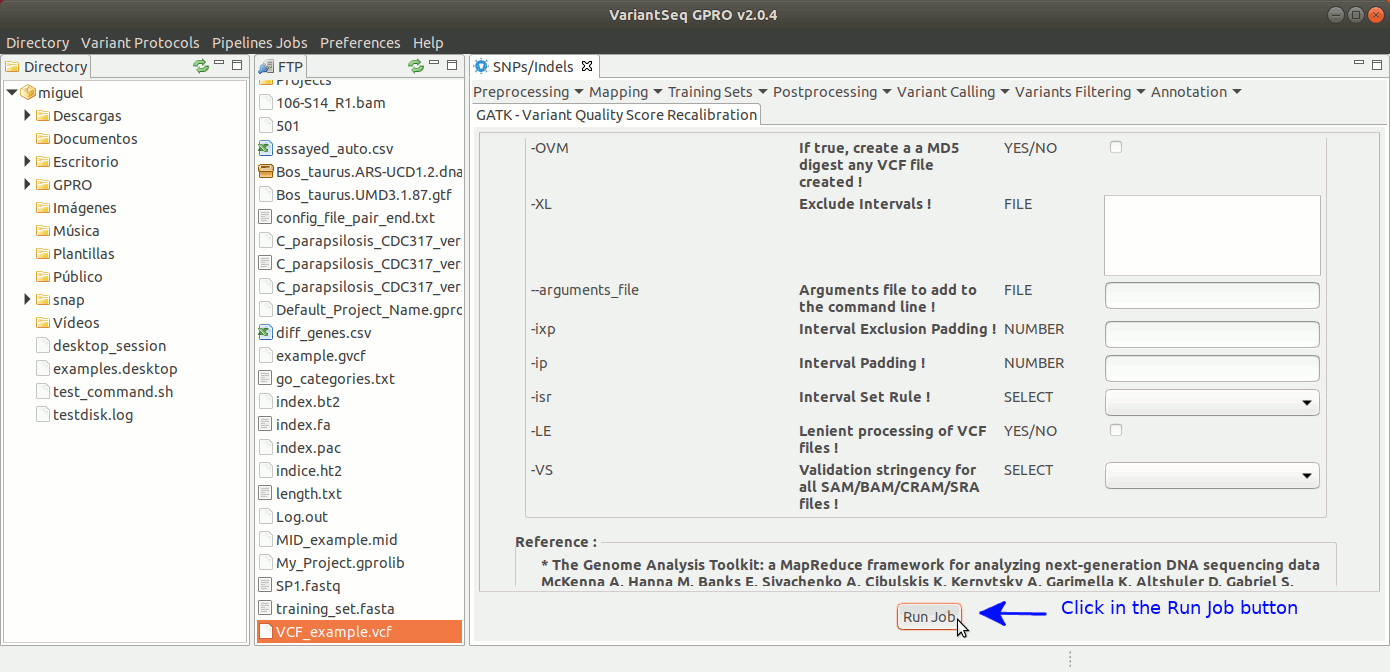
Figure 39: Using the GPRO interface for VQSR.
- Variant Filtering : Variant Filtration
Variant Filtration is a GATK utility for hard filtering of variant calls based on INFO and FORMAT annotations of the VCF file. For more details, see the GATK Forum
To run Variant Filtration, go to [ Variant Filtering → Variant Filtration ] and follow Fig. 40.
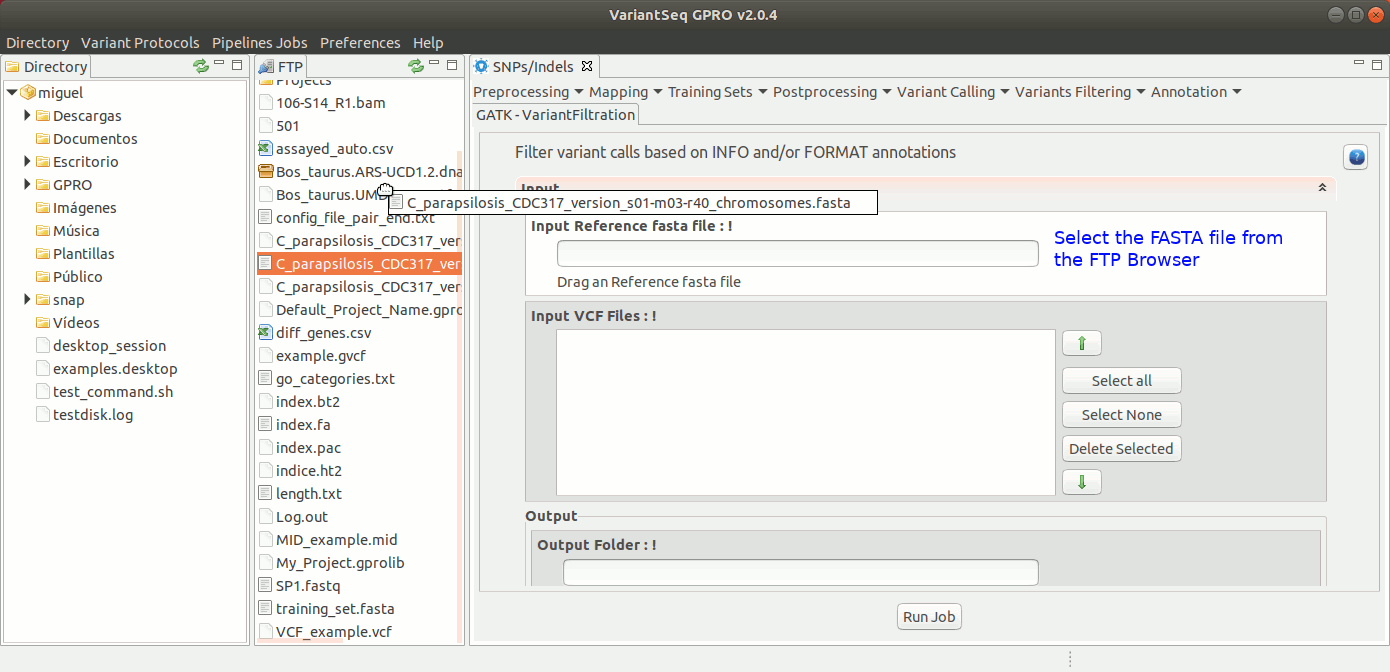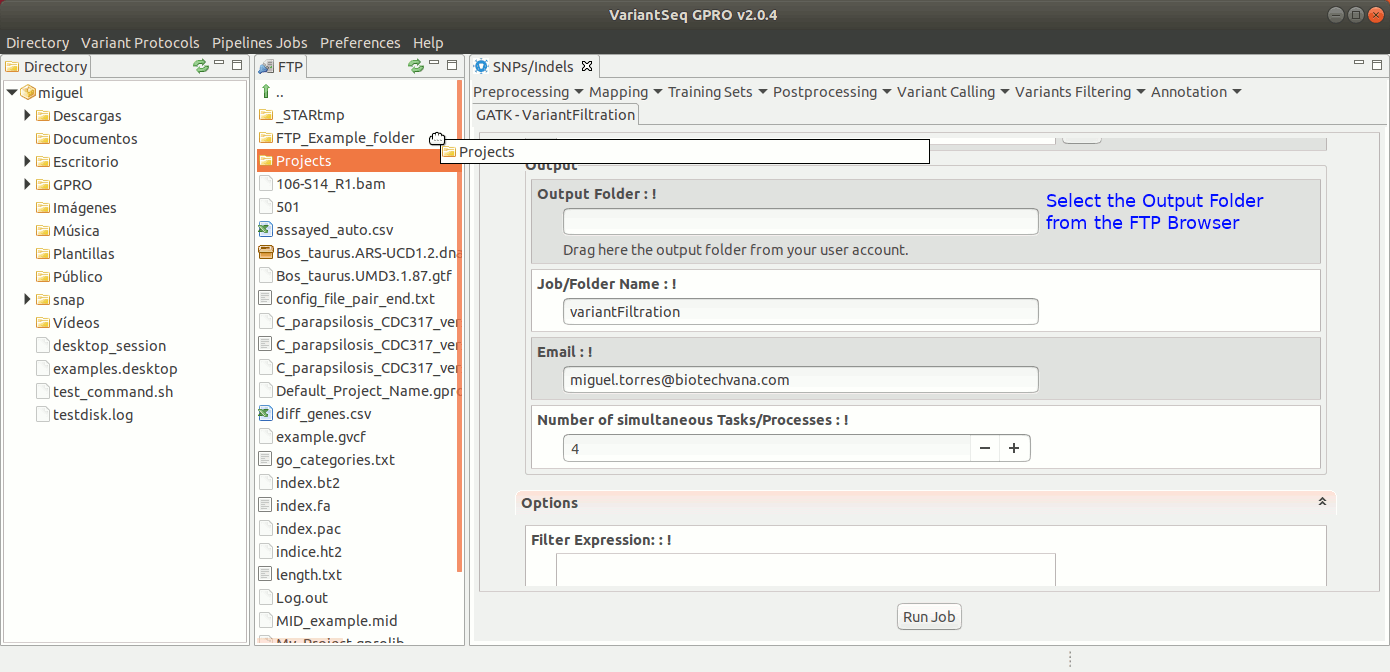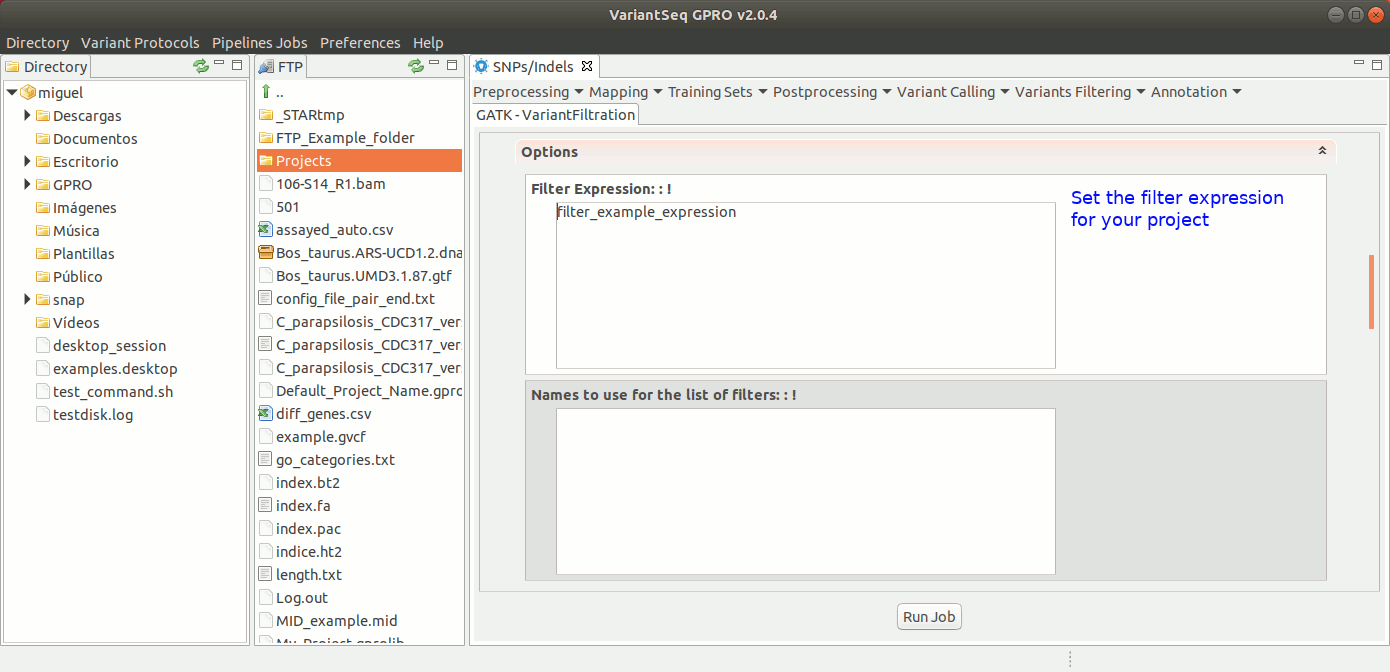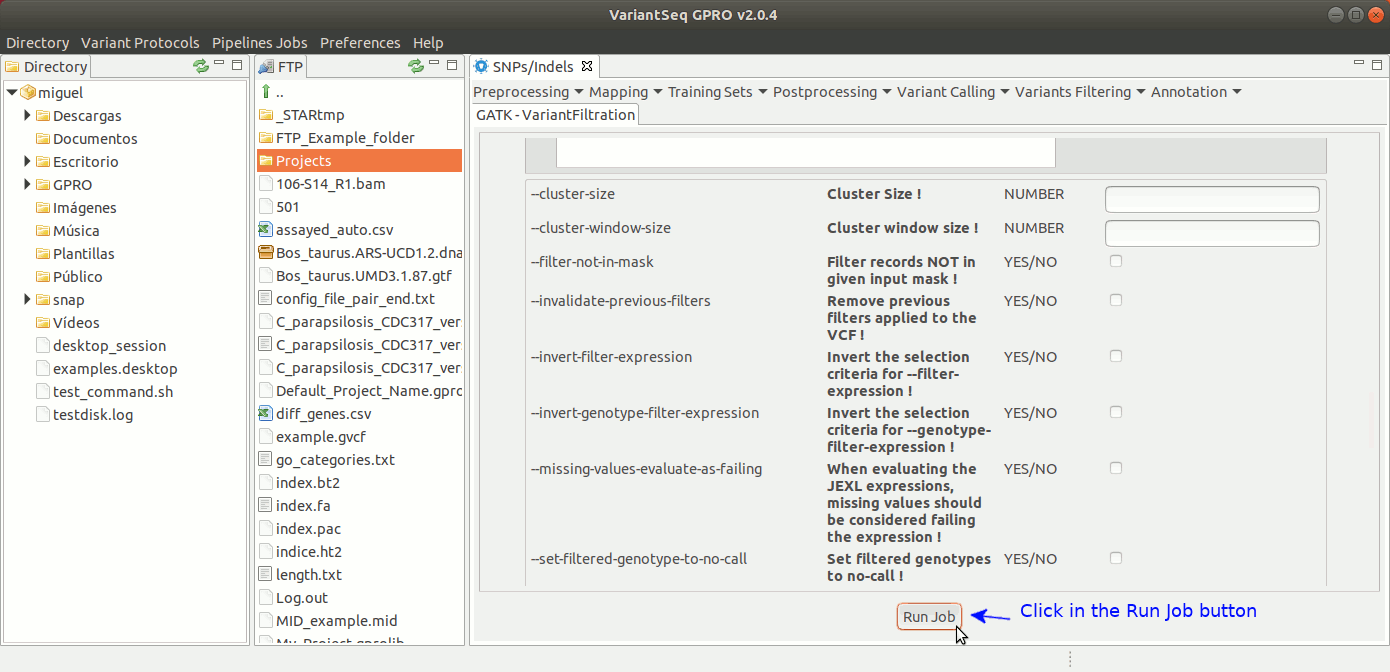
Figure 40: Using the GPRO interface for variantFiltration.
- Variant Filtering : Cross-sample contamination
Cross-sample contamination applies for cancer projects using ContEst a GATK tool that determines the percent contamination in tumor samples (by sample, by lane, or in aggregate across all the input reads) in comparison to their normal pairs (it only works for tumour-normal pairs). Cross-sample contamination allows you to also include VCF files with population allele frequencies derived from the gnomAD resource. For more details, see the GATK Forum
To run Cross-sample contamination, go to [ Variants Filtering → GATK Cross-sample contamination ] and follow Fig. 41.
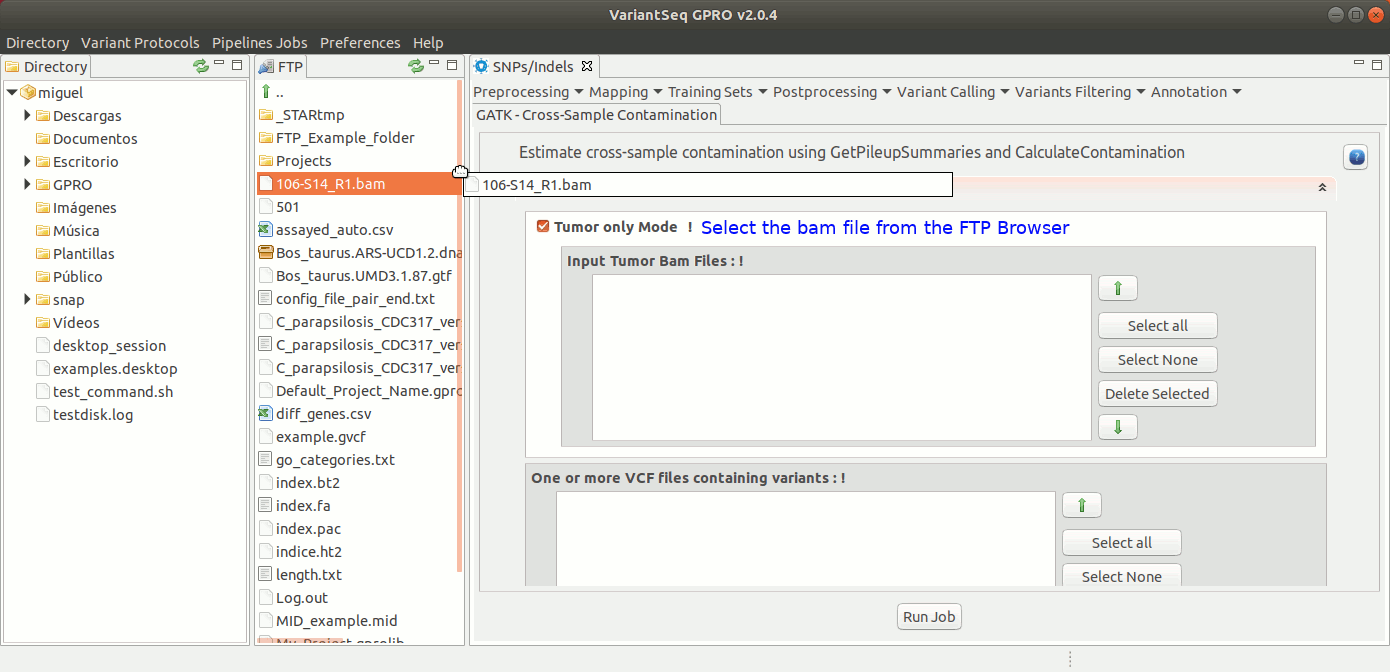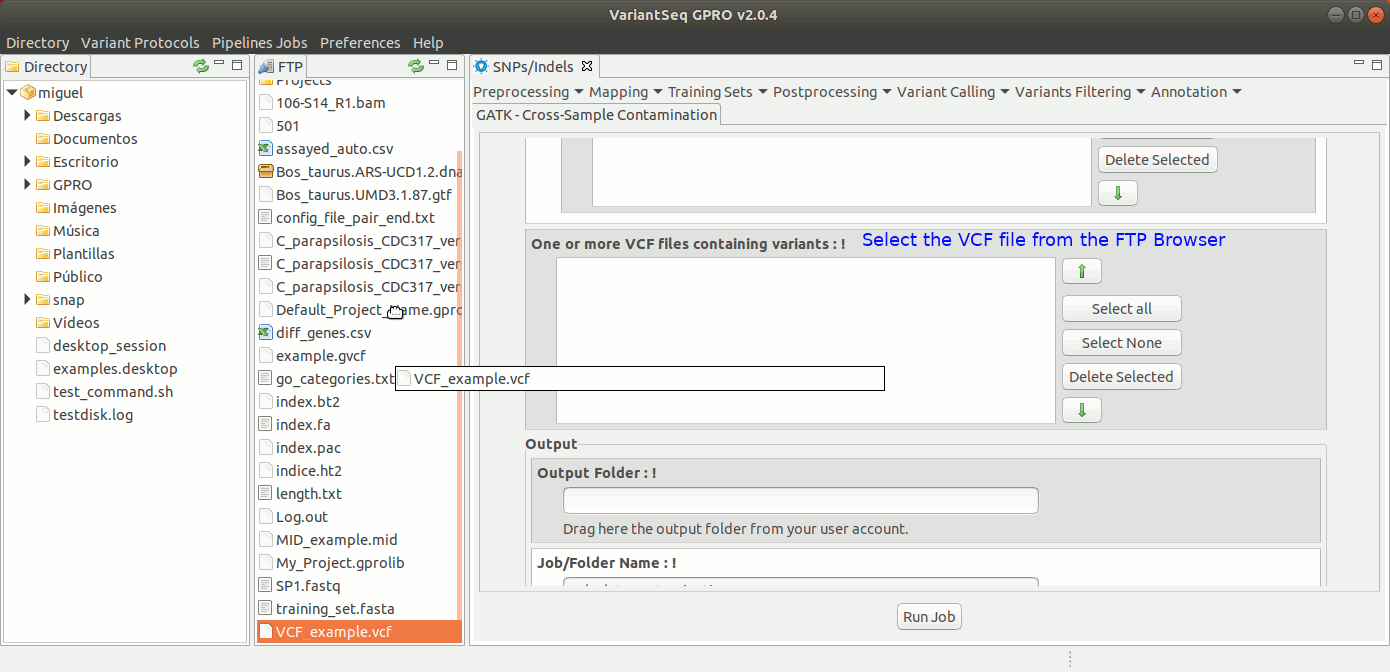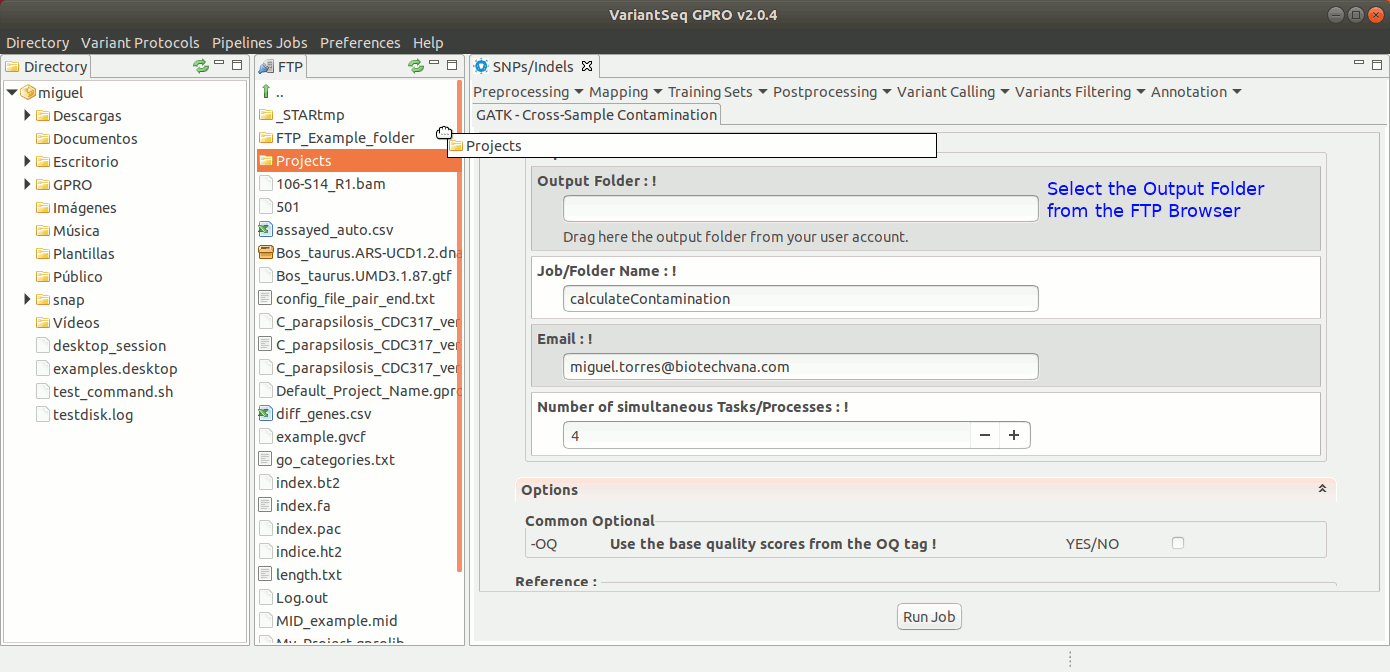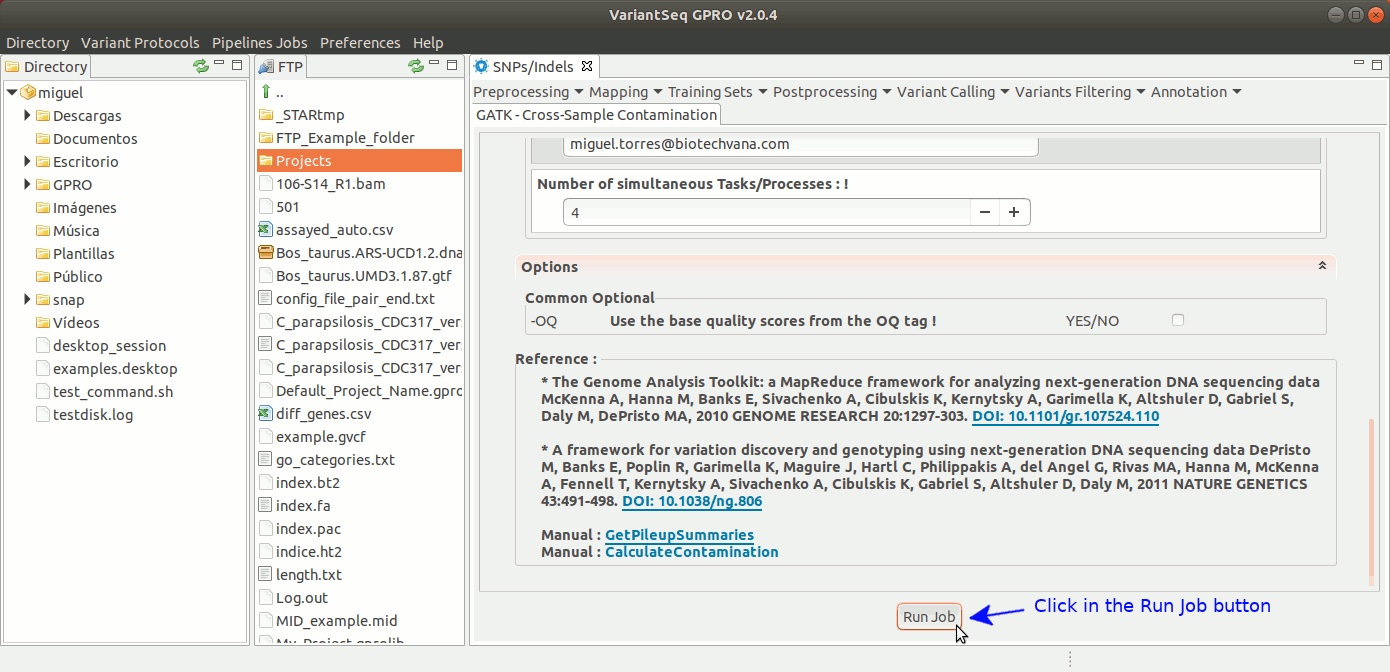
Figure 41: Using the GPRO interface for Cross-sample contamination.
- Variant Filtering: FilterMutectCalls
FilterMutectCalls is a GATK beta tool of Mutect2 to filter somatic SNPs and indels. For more details, see the GATK Forum
To run FilterMutectCalls, go to [ Variant Filtering → FilterMutectCalls ] and follow Fig. 42.
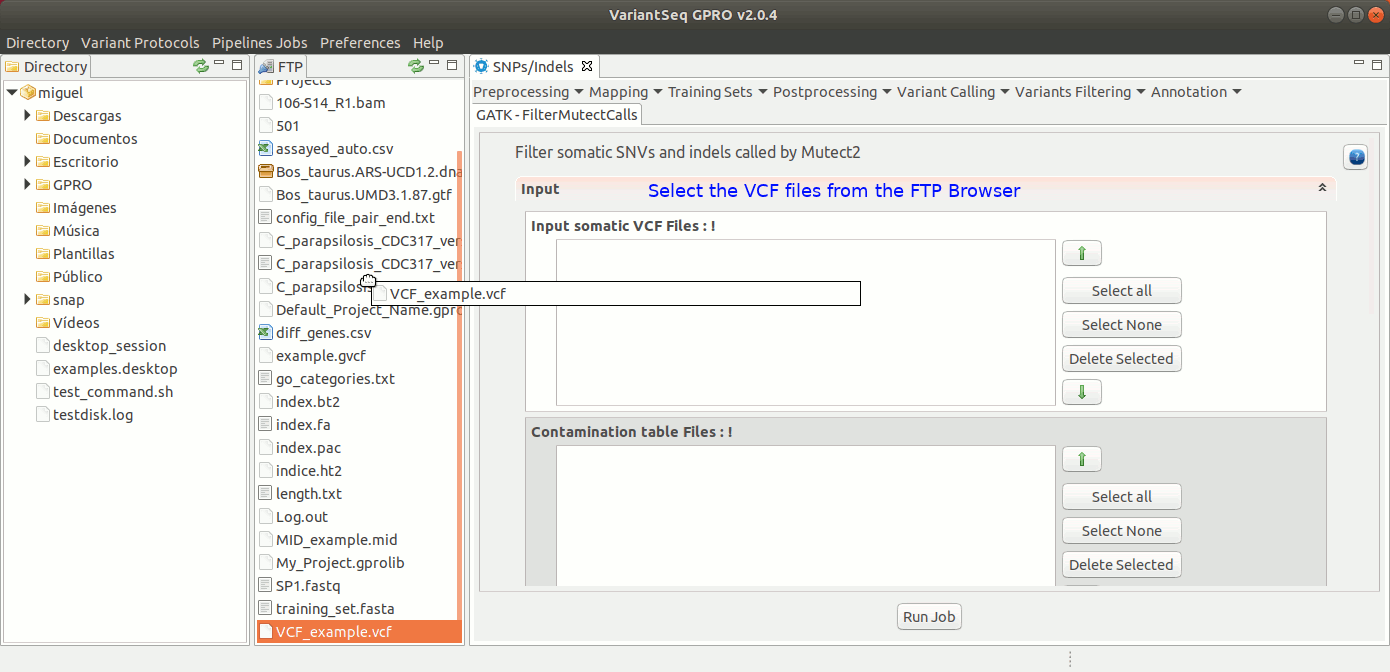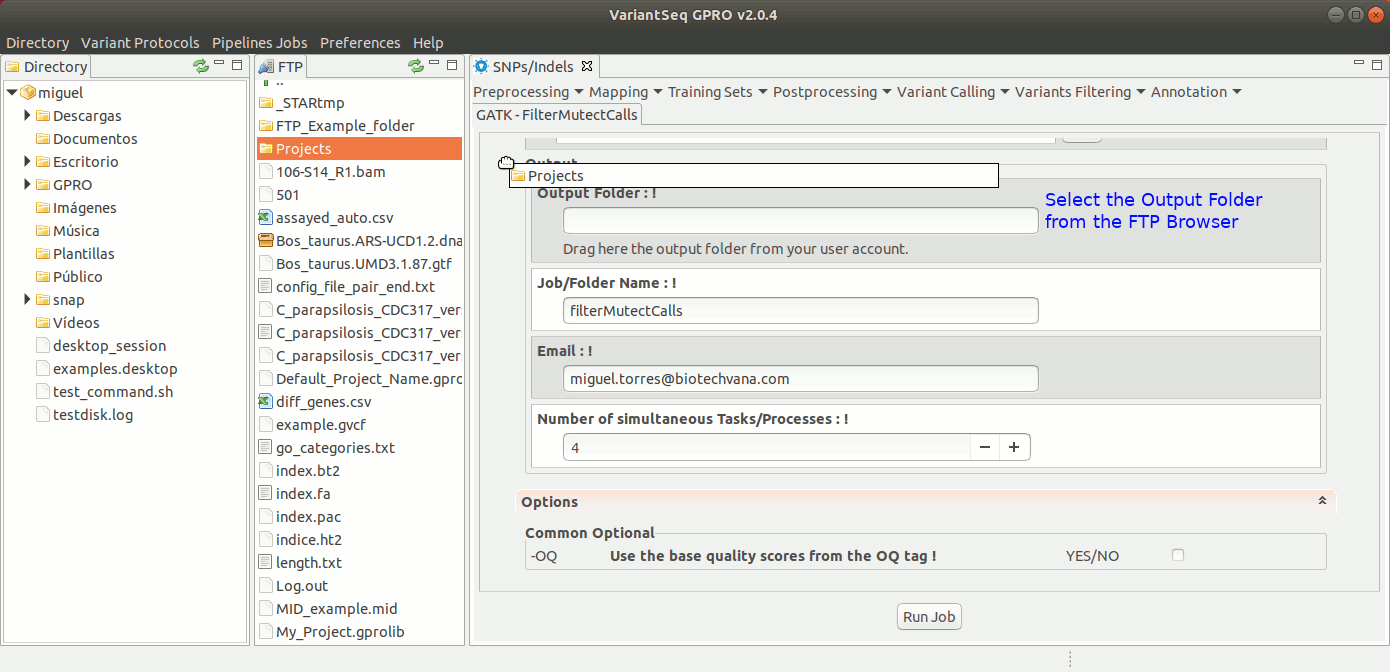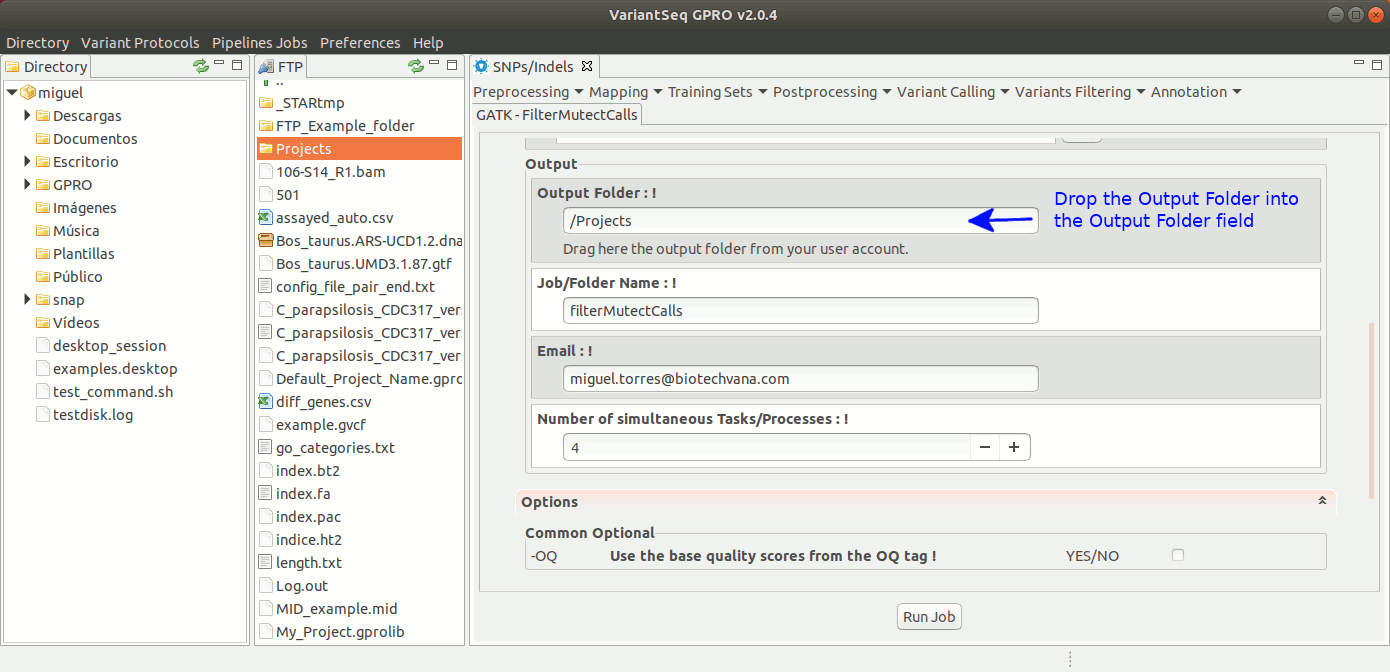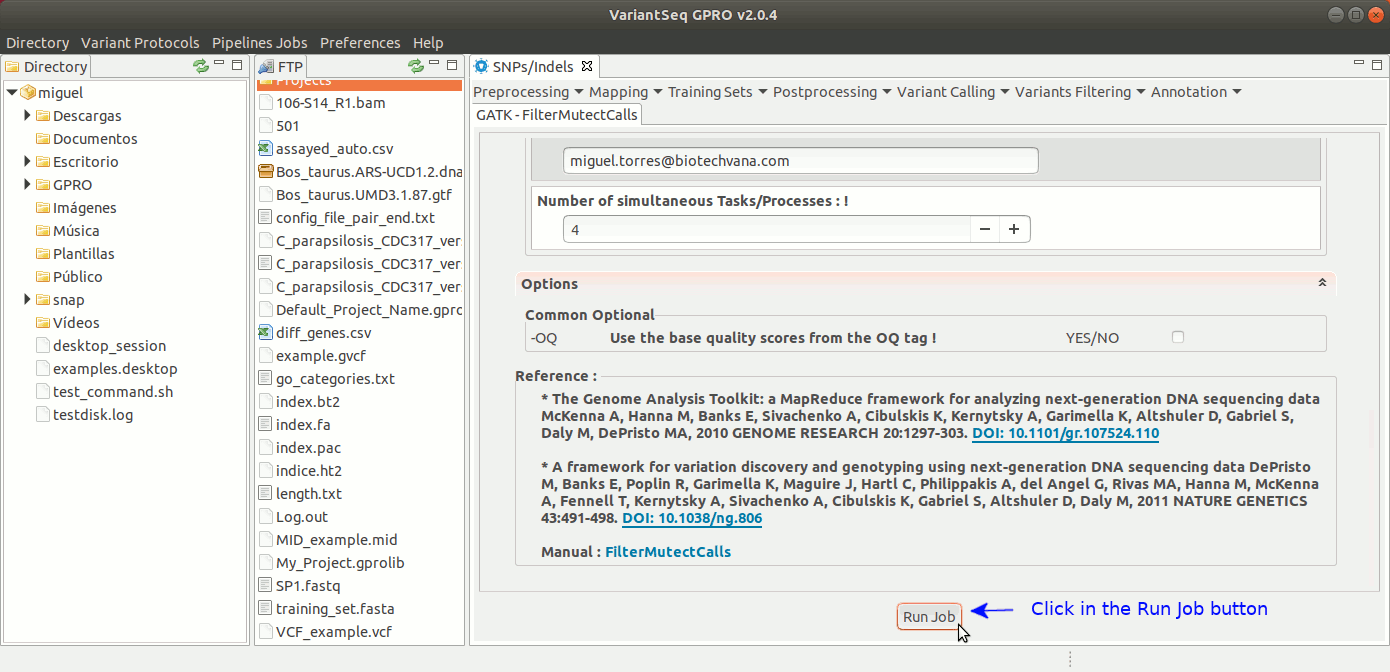
Figure 42: Using the GPRO interface for FilterMutectCalls.
To annotate SNPs and Indels effects, VariantSeq implements an interface to execute the Variant Effect Predictor (VEP) of Ensembl (McLaren et al. 2016). This provides annotations for the genes and transcripts affected by the variants, the location of the variants, their functional consequences on the protein sequence (e.g. stop gained, missense, stop lost, frameshift), information about known variants that matching your gene. It also provides annotations for minor allele frequencies from 1000 Genomes Project, and SIFT and PolyPhen-2 scores for changes to protein sequence. For more details about VEP, see: https://www.ensembl.org/info/docs/tools/vep/index.html
To execute Ensembl VEP. go to [ Annotation → Variant Effect Predictor ] and follow Fig. 43.
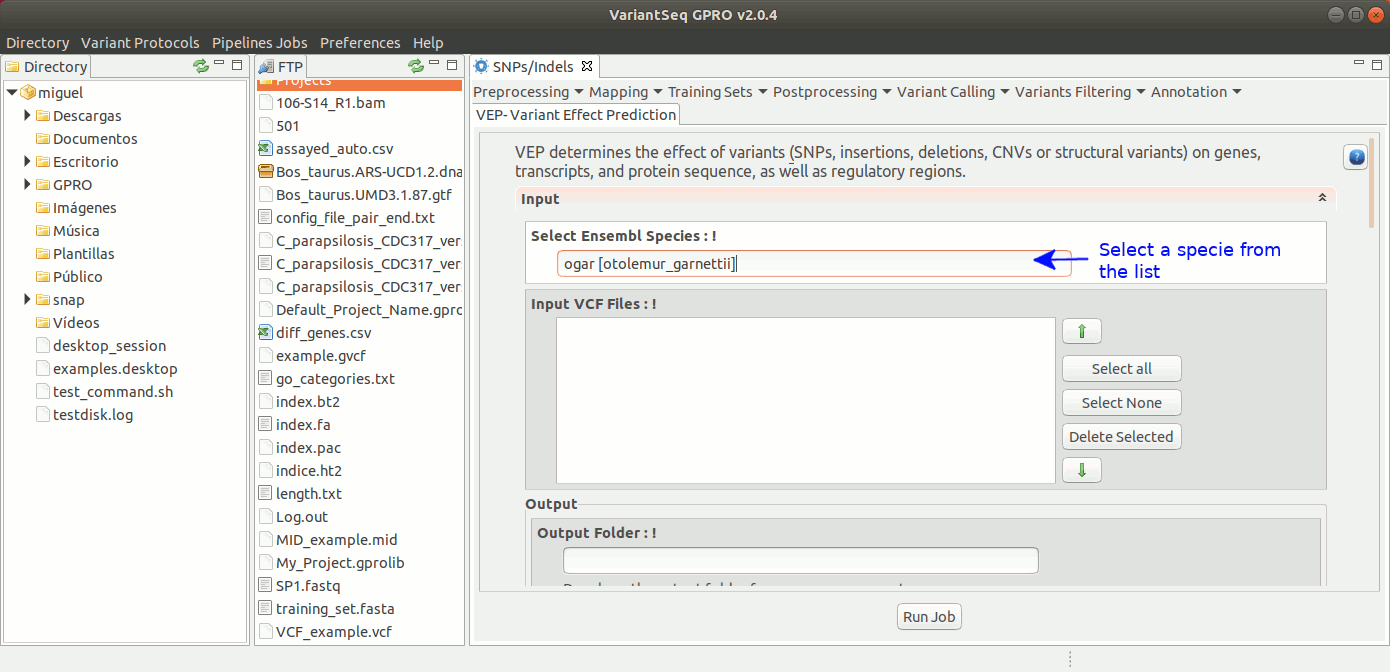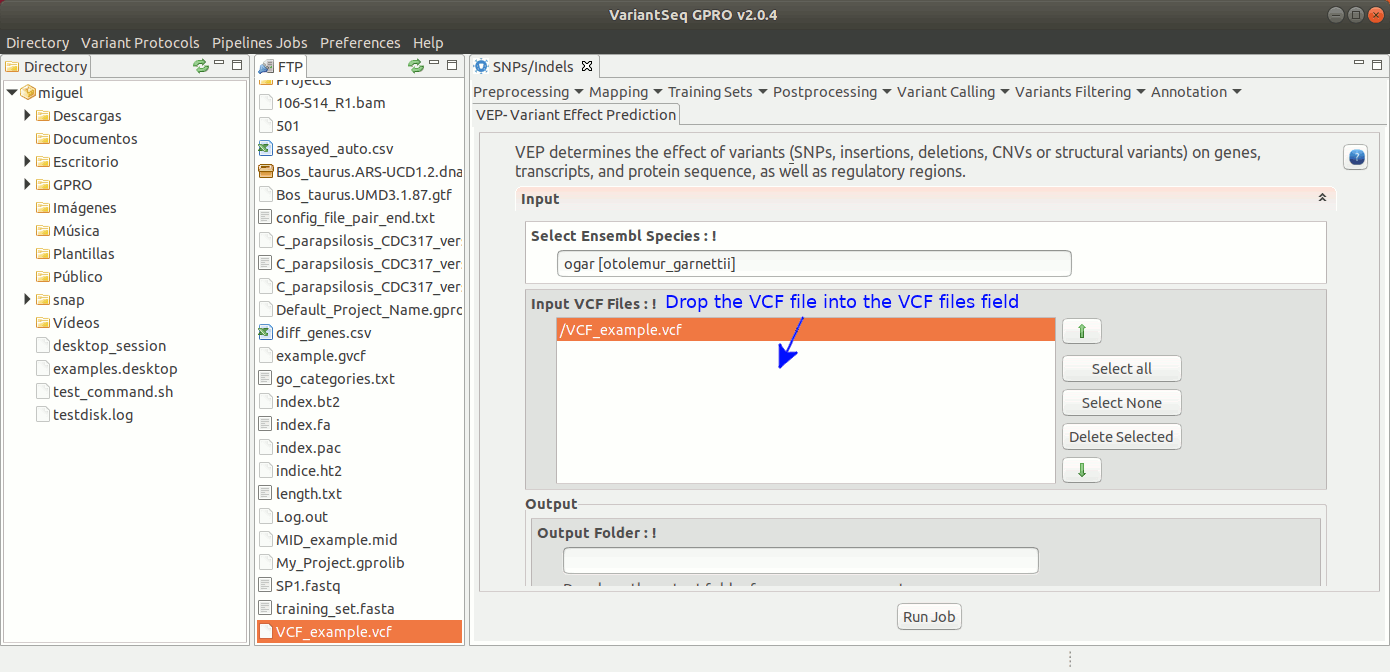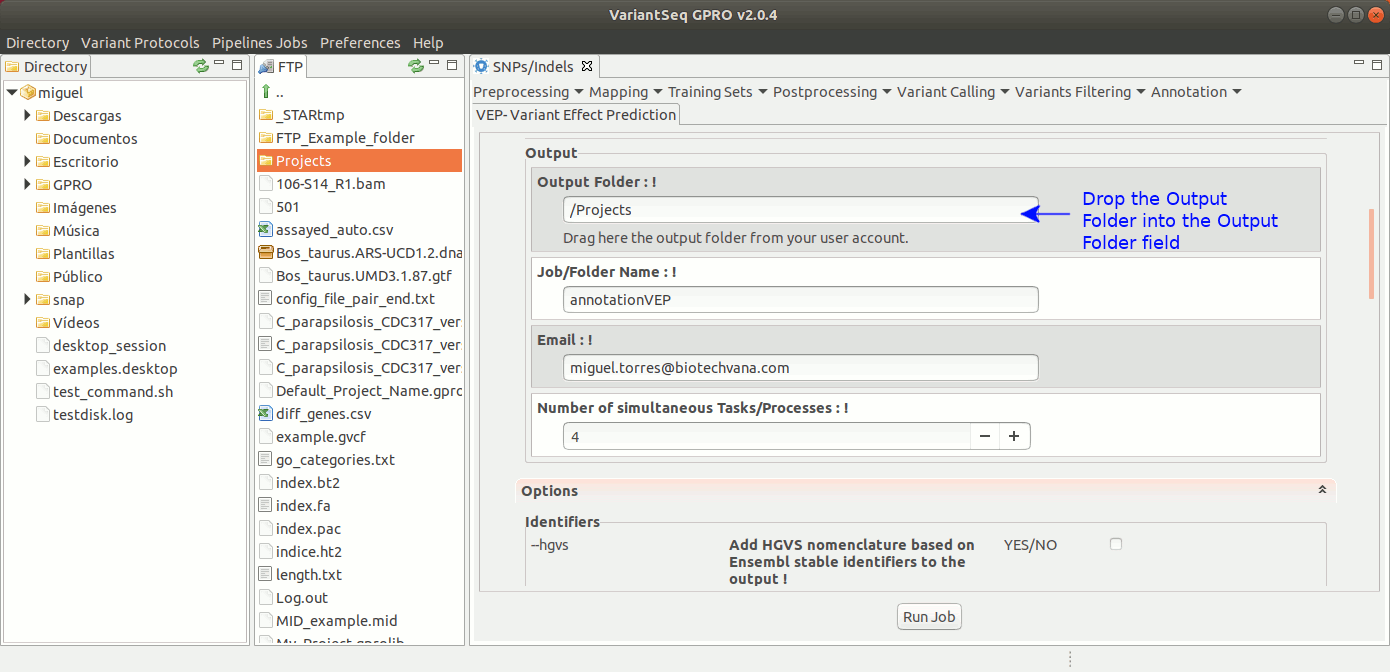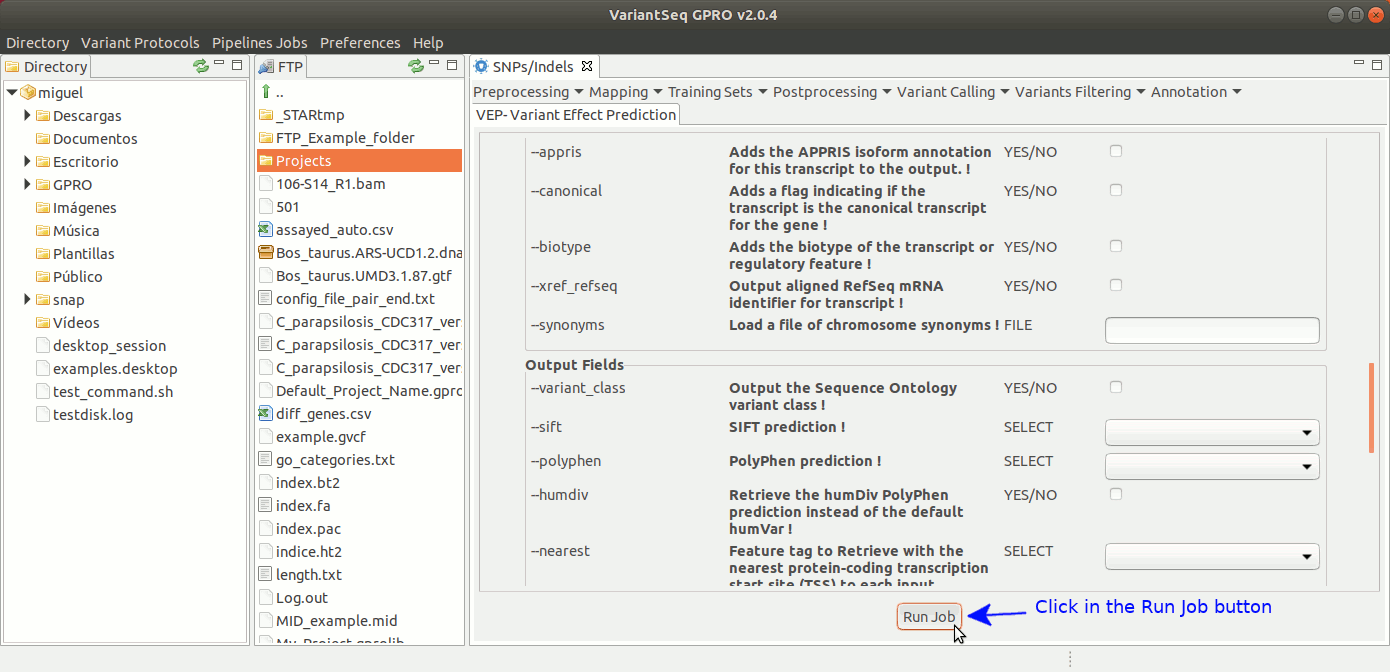
Figure 43: Using the GPRO interface for VEP.







