[Transcripts Protocols → Step-by-Step → Tophat/Hisat2 & Cufflink Protocol]
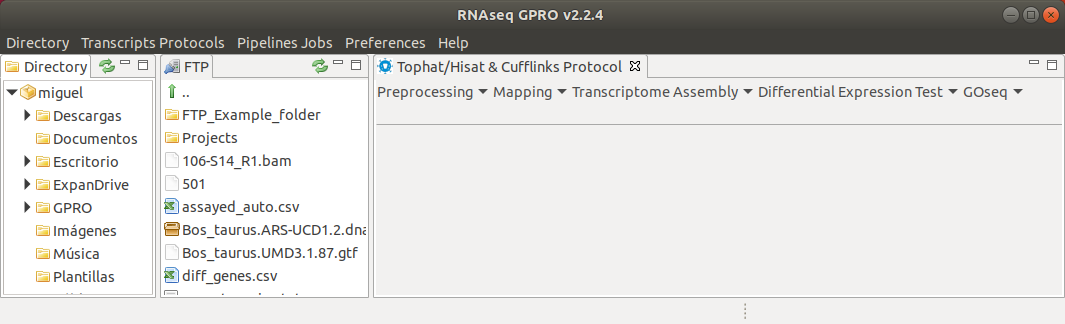
Figure 5: Submenu to follow the Tophat & Cufflinks protocol in Step-to-step mode.
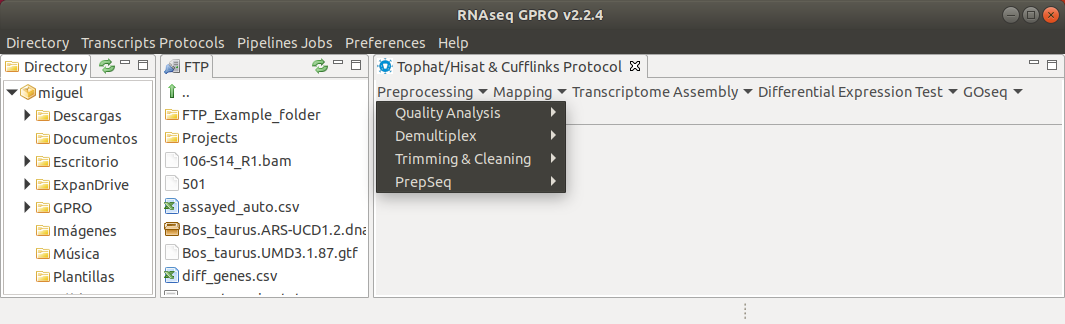
Figure 6: Preprocessing drop down submenu listing the preprocessing options enabled in the RNA application.
- Quality Analysis: FastQC
FastQC (Andrews 2016) provides a simple way to perform quality checks on raw sequence data.FastQC provides a modular set of analyses that can indicate whether your data contain potential artifacts that require “cleaning” before beginning any analyses. Upon performing a FastQC quality check, you will obtain a complete sequence quality report provide hints on what form of filtering and processing your sample requires if any. For example, overrepresented sequences that correspond to the adapters used during sequencing may need to be removed from your fastq files. This will be shown in the “overrepresented sequences” section of the FastQC report. Further details on FastQC can be found in the FastQC manual at https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
To run FastQC go to [ Preprocessing → Quality Analysis → FastQC ] and follow Fig. 7
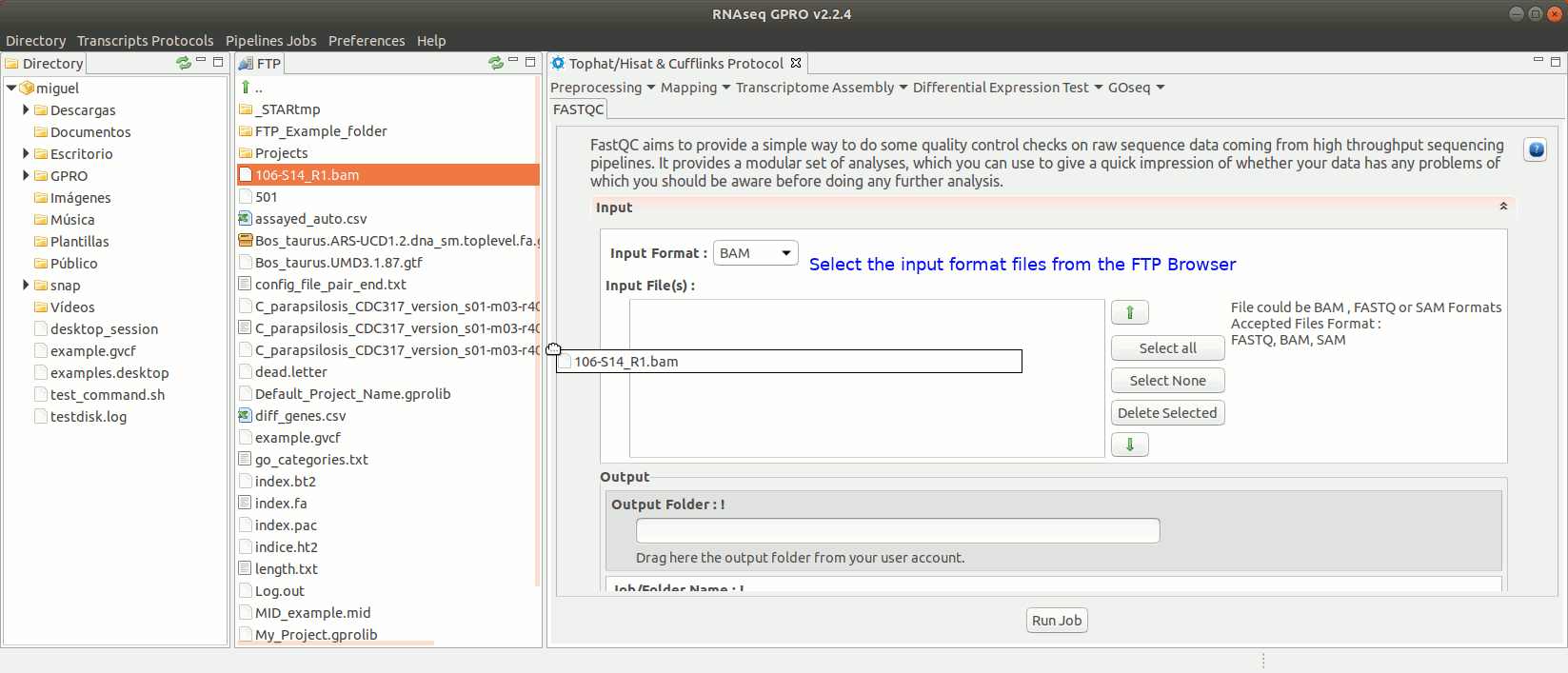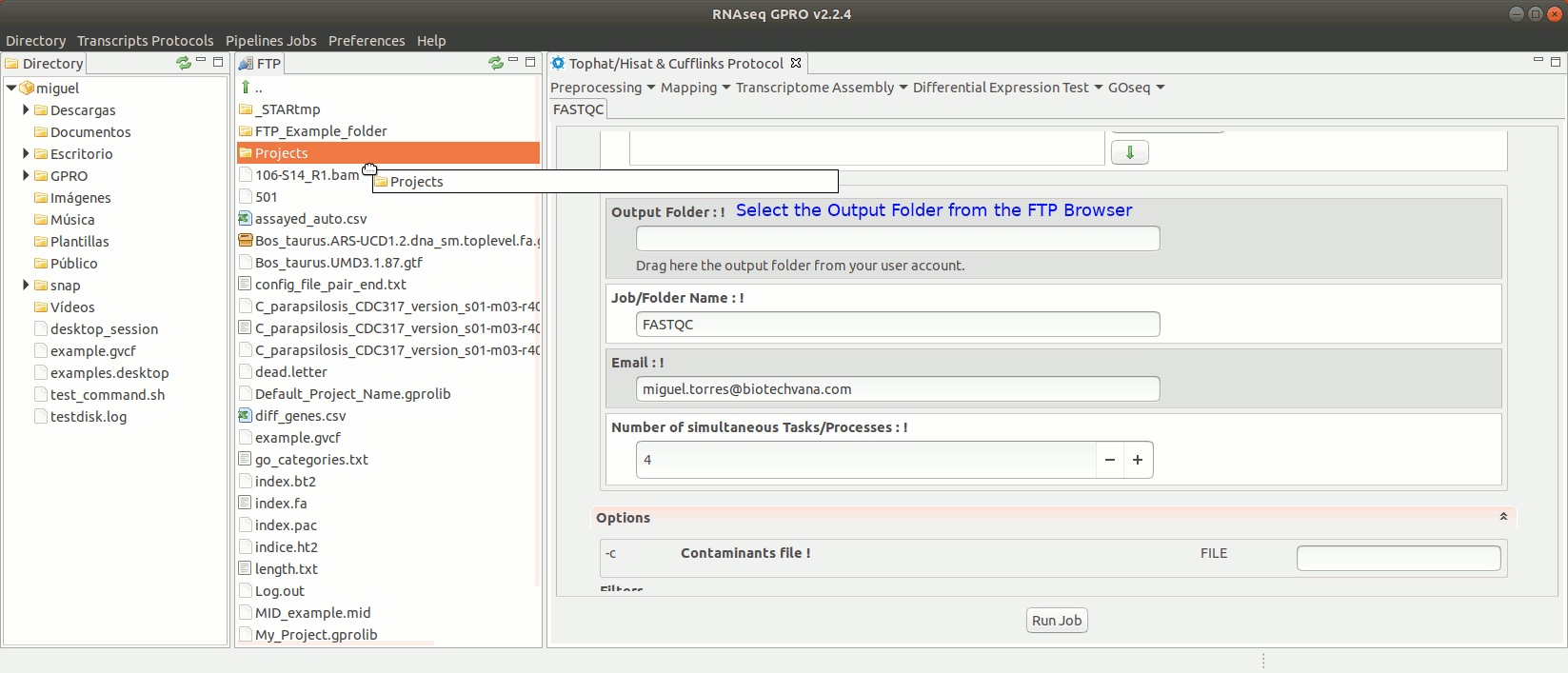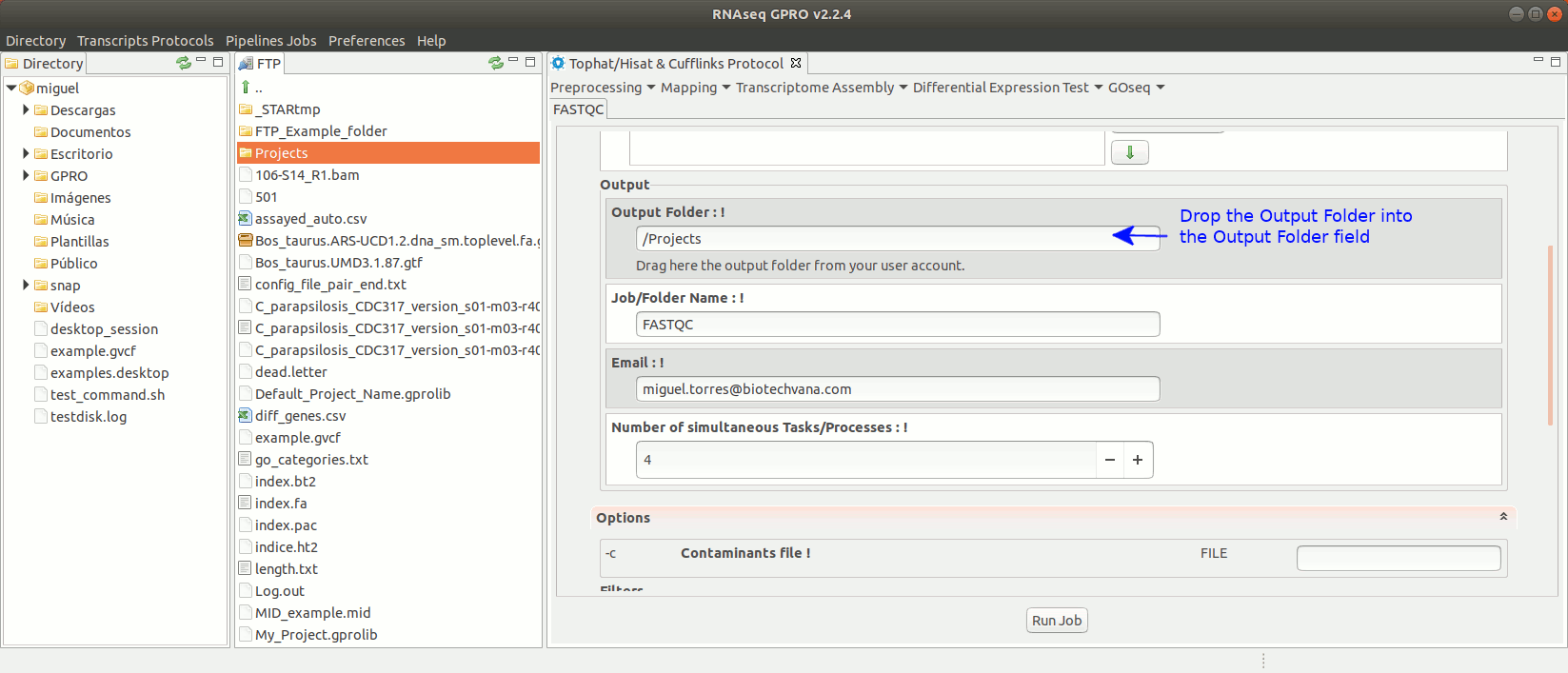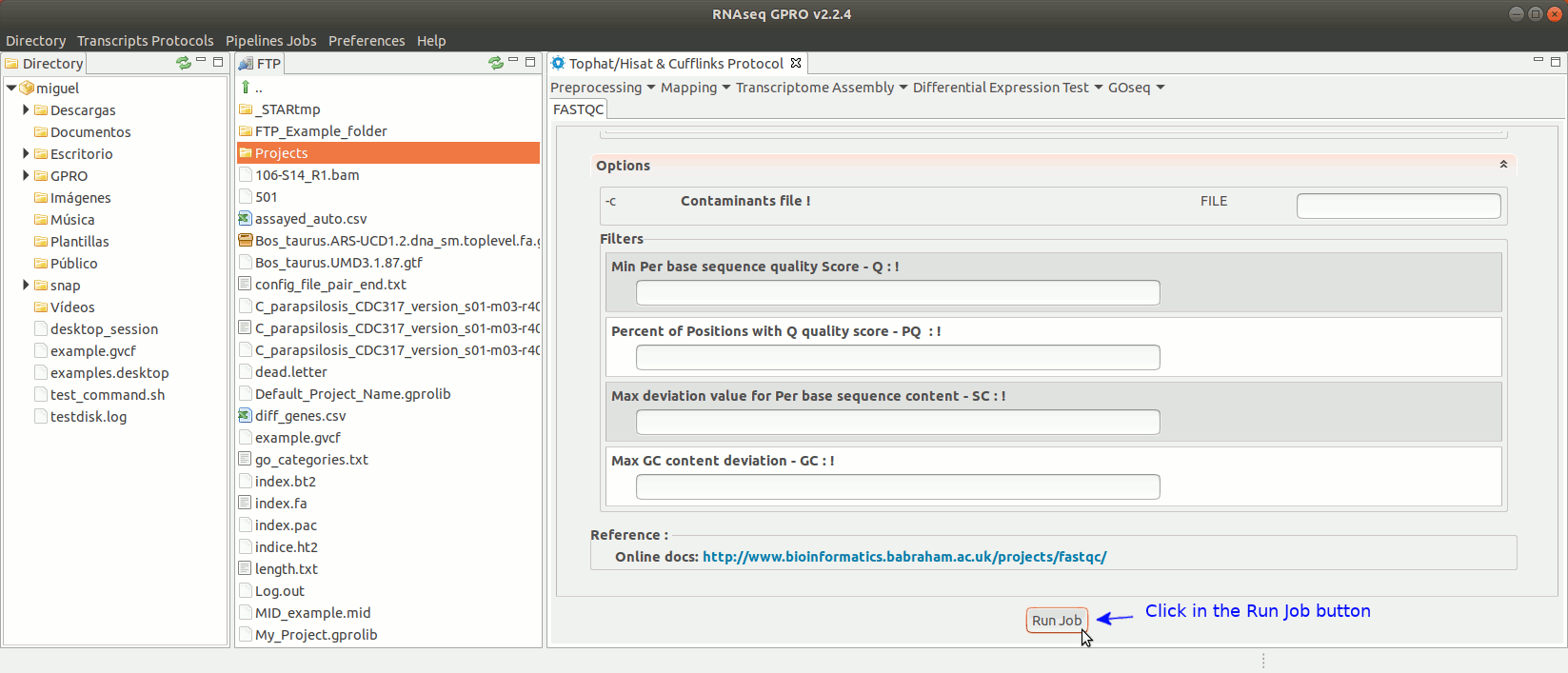
Figure 7: Using the GPRO interface for FastQC.
- Demultiplex: FastqMidCleaner
FastqMidCleaner sorts and splits sequencing reads from fastq files into separate files according to predefined molecular identifiers (MIDs).
To run FastqMidCleaner go to [Preprocessing→Demultiplex → FastqMidCleaner] and follow Fig. 8.
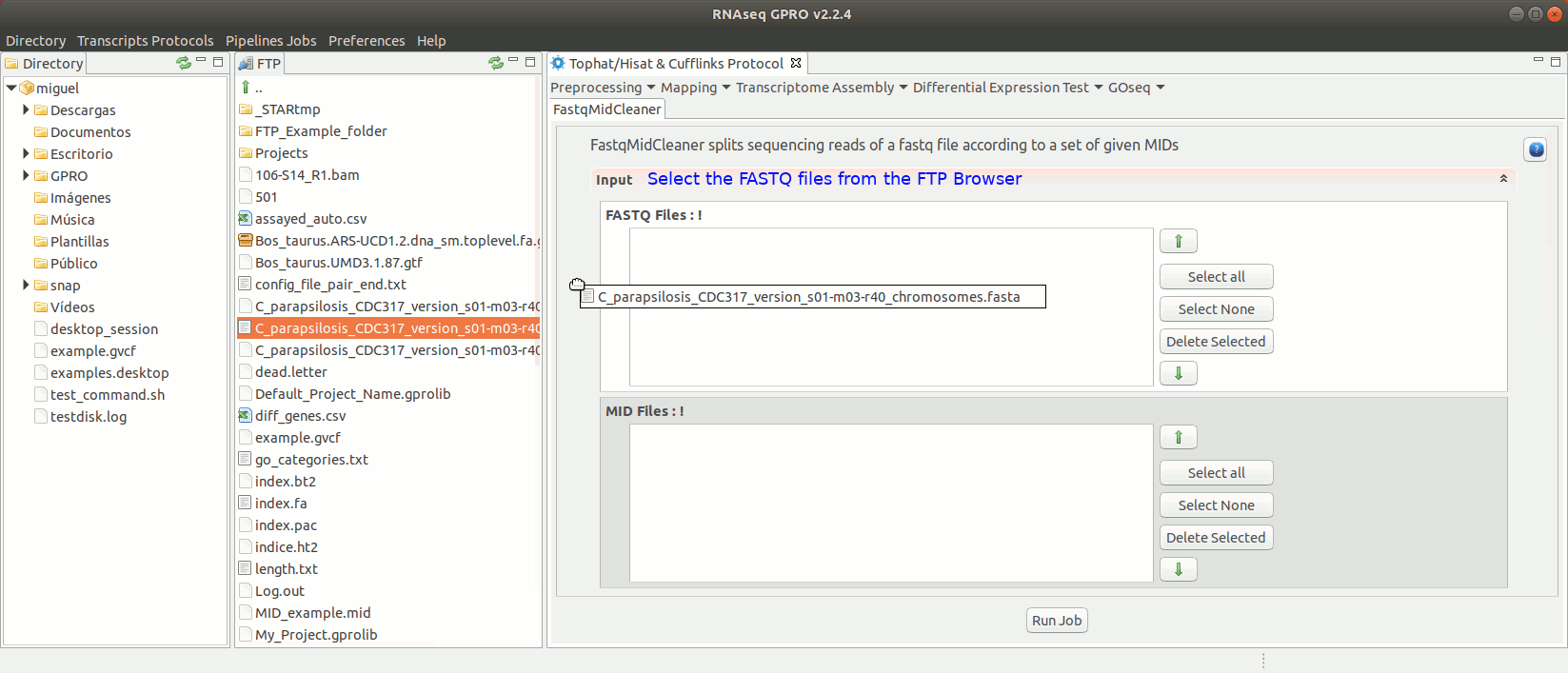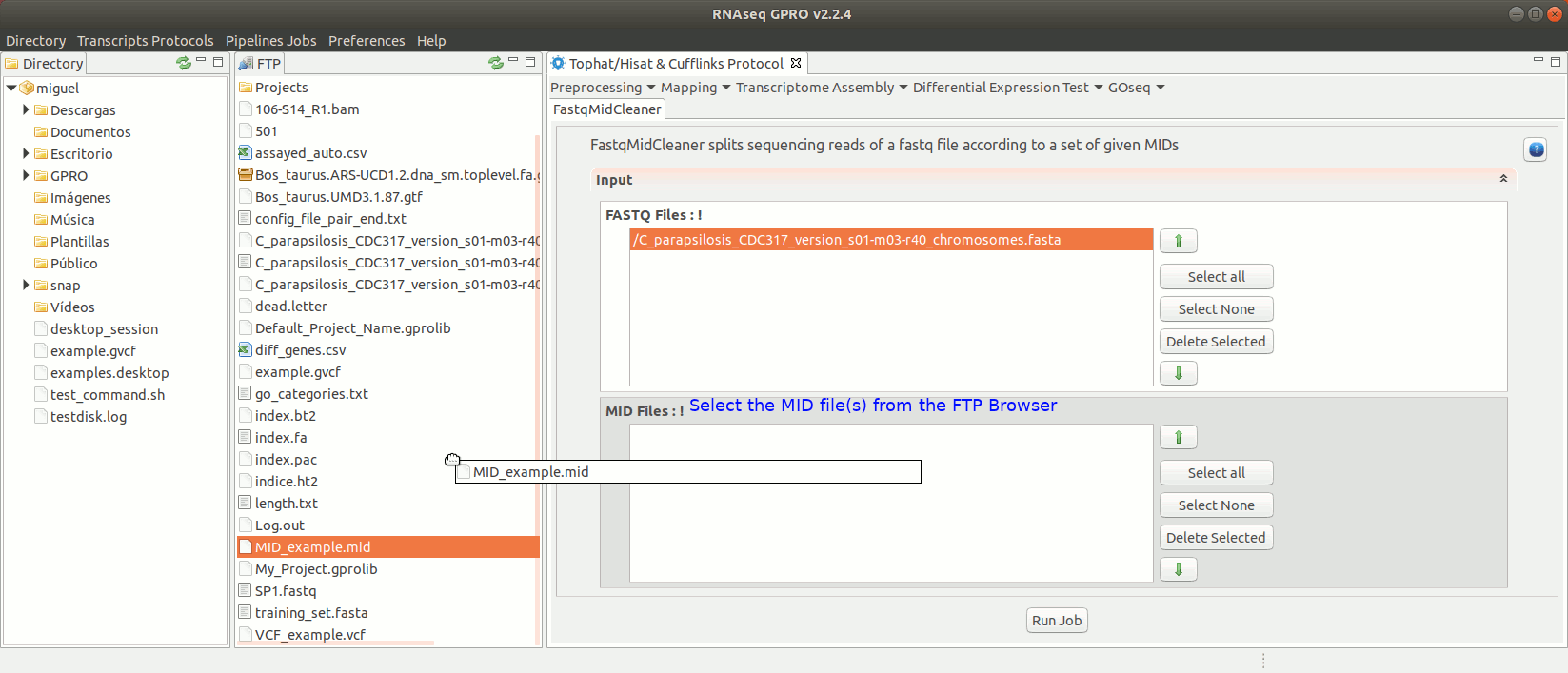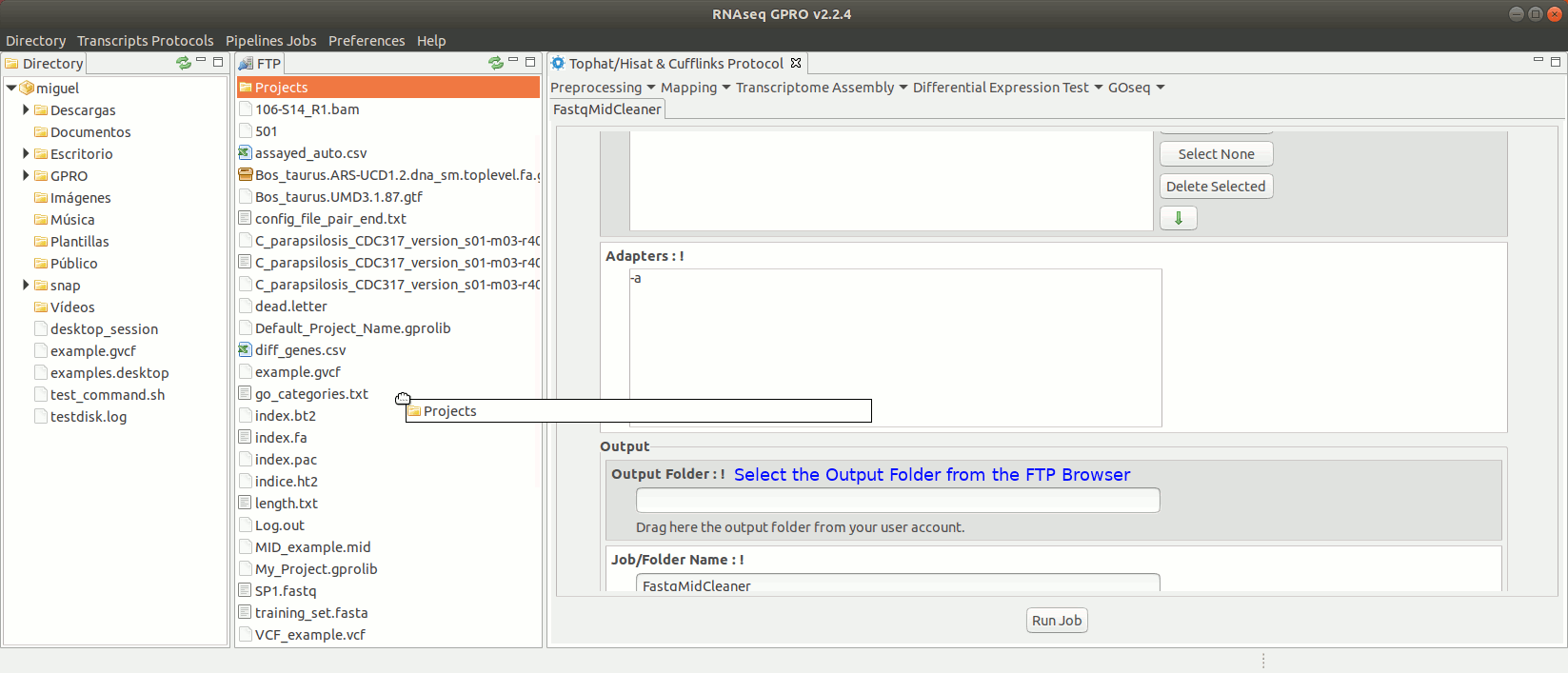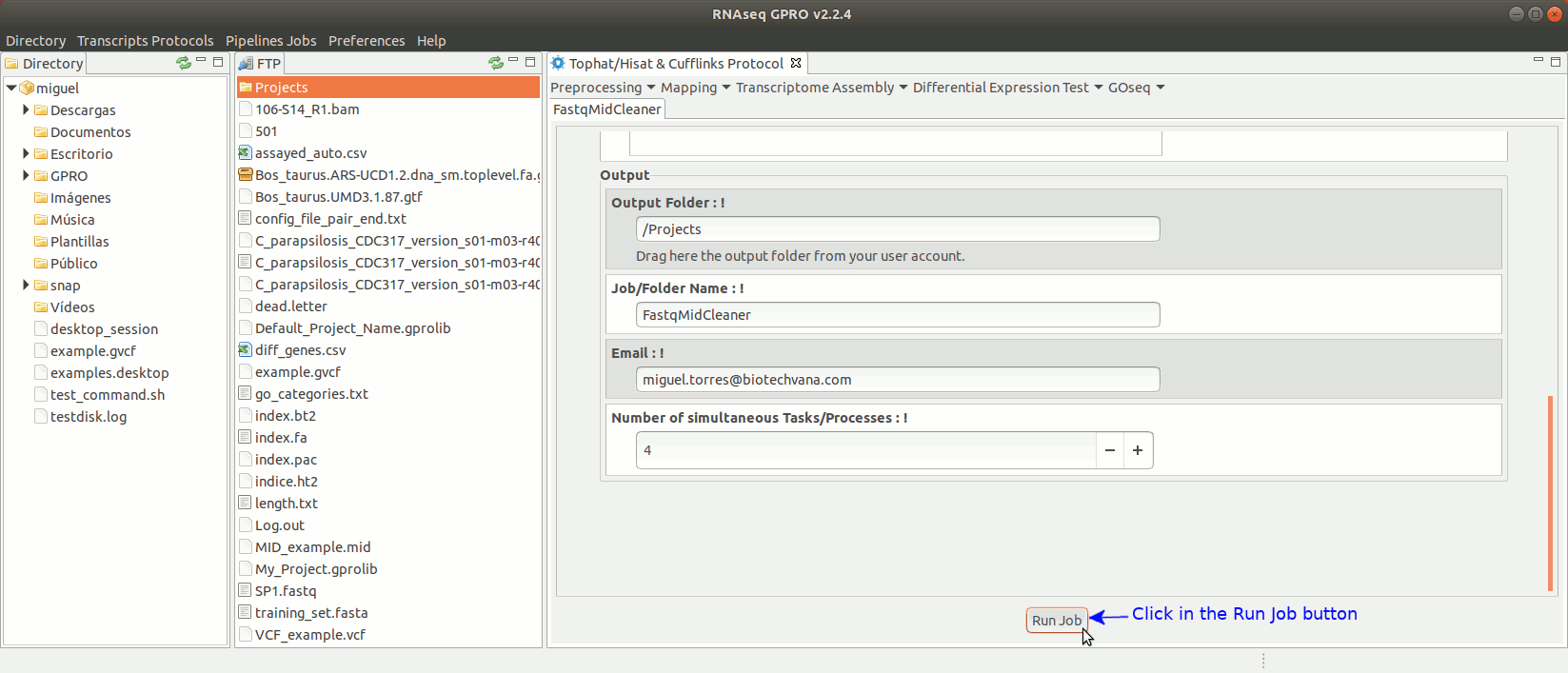
Figure 8: Using the GPRO interface for FastQMidCleaner.
Cutadapt (Martin 2011) finds and removes adapter sequences, primers, poly-A tails and other types of unwanted sequences from your sequencing reads. For more information on Cutadapt, see its manual at https://cutadapt.readthedocs.io/en/stable/guide.html
To run Cutadapt go to [ Preprocessing→ Trimming & Cleaning → Cutadapt ] and follow Fig. 9.
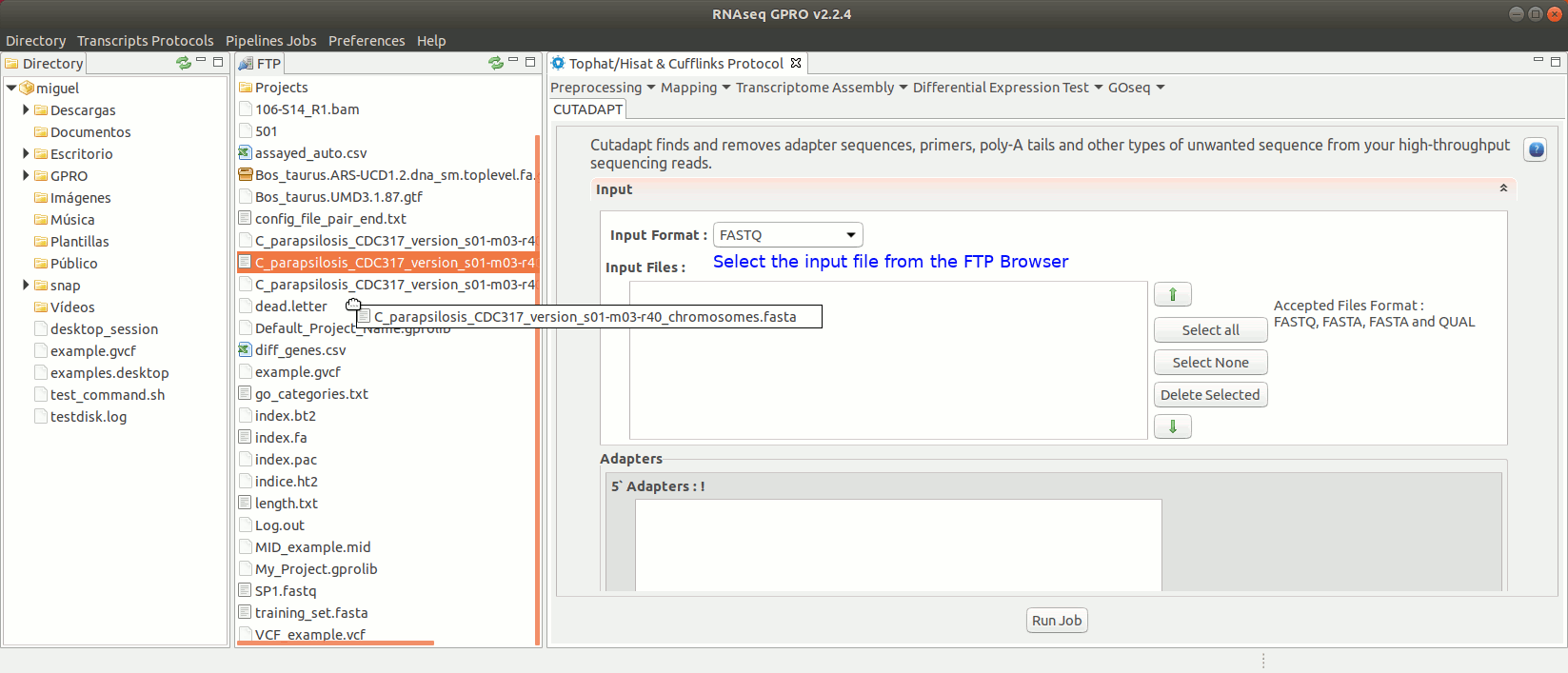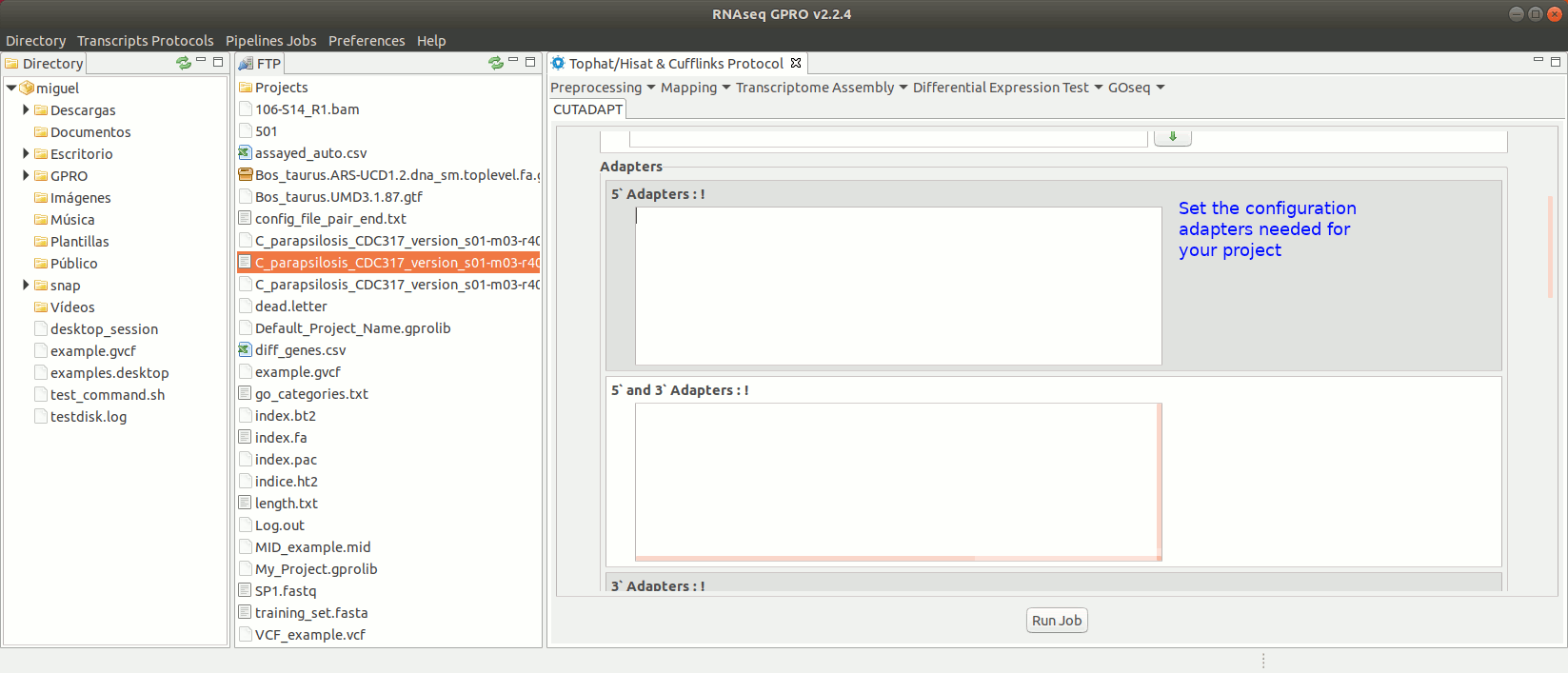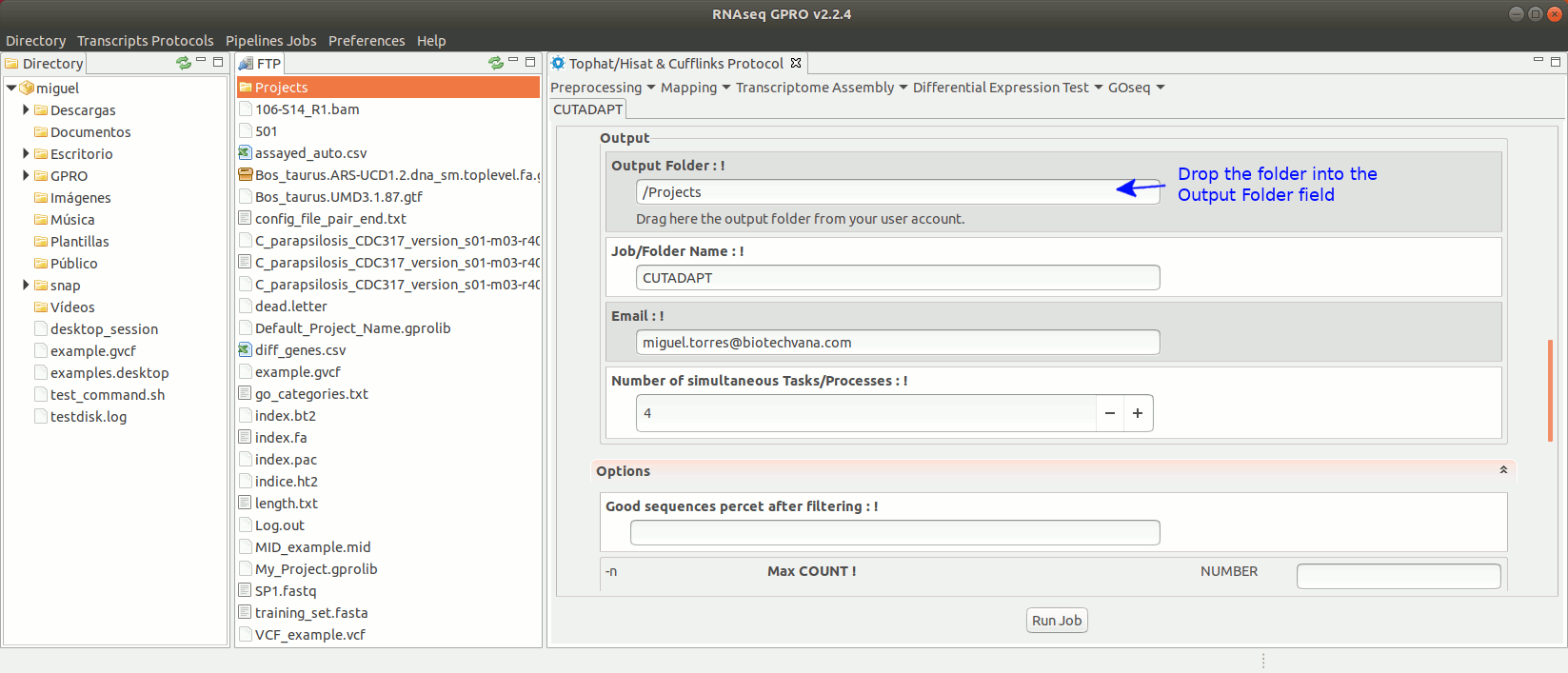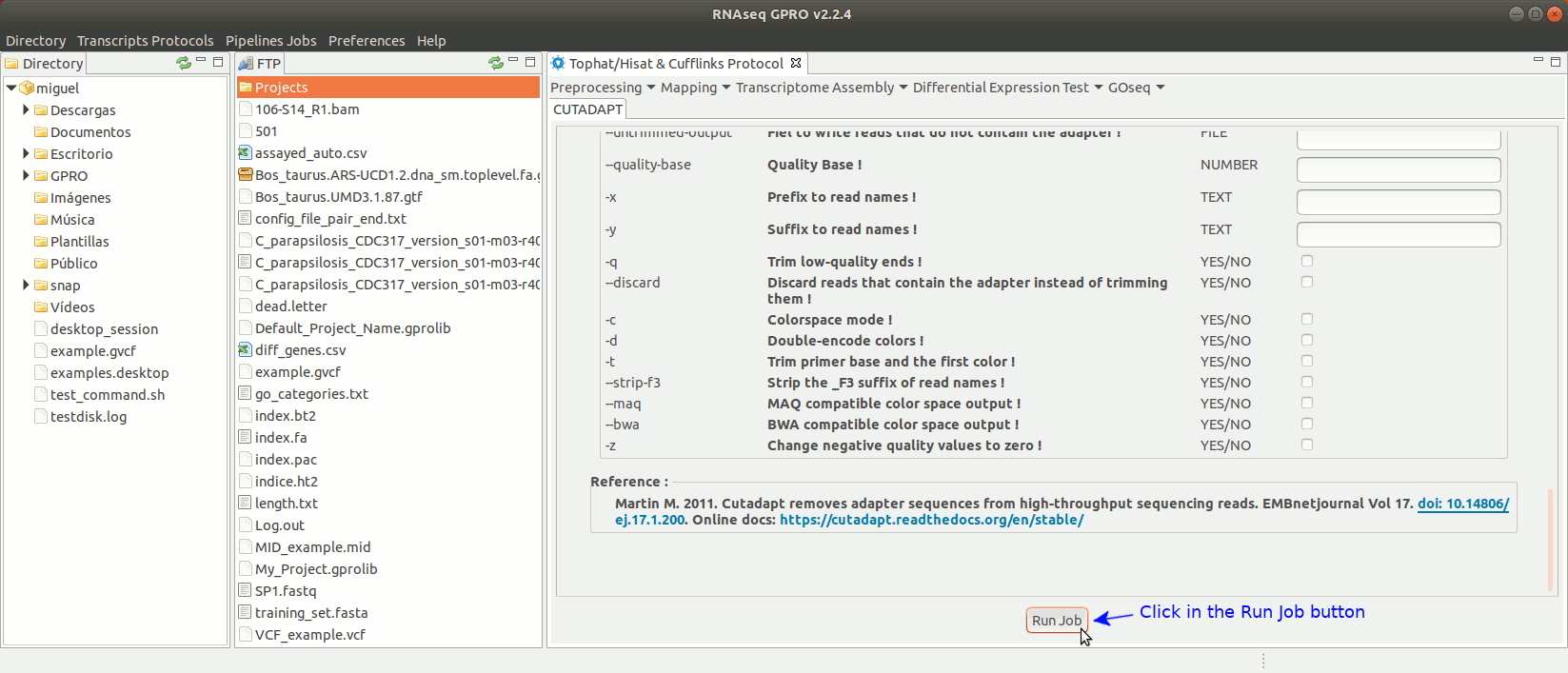
Figure 9: Using the GPRO interface for Cutadapt.
- Trimming & Cleaning: Prinseq
Prinseq (Schmieder and Edwards 2011) can be used to filter, reformat, or trim your sequencing reads. For further information see Prinseq manual at http://prinseq.sourceforge.net.
To run Prinseq go to [Preprocessing→Trimming & Cleaning → prinseq ] and follow Fig. 10.
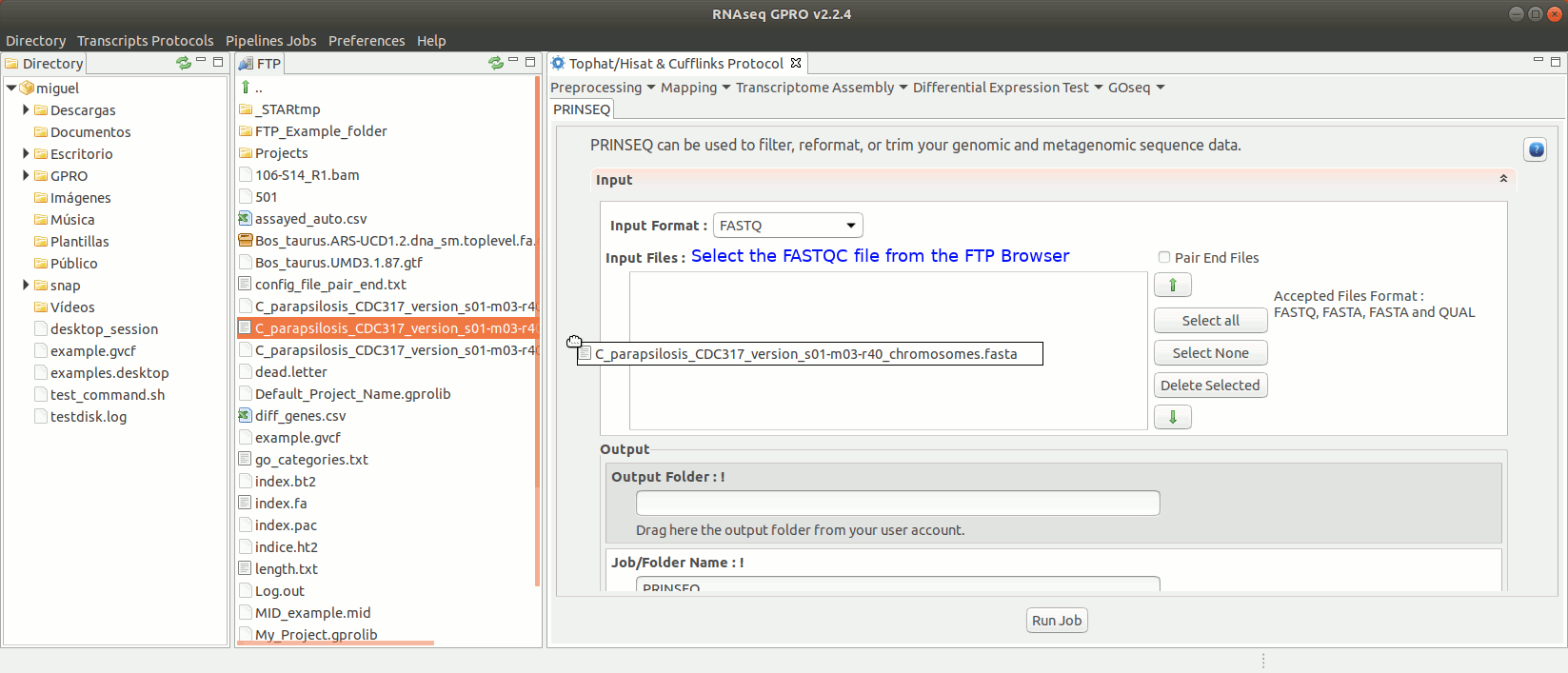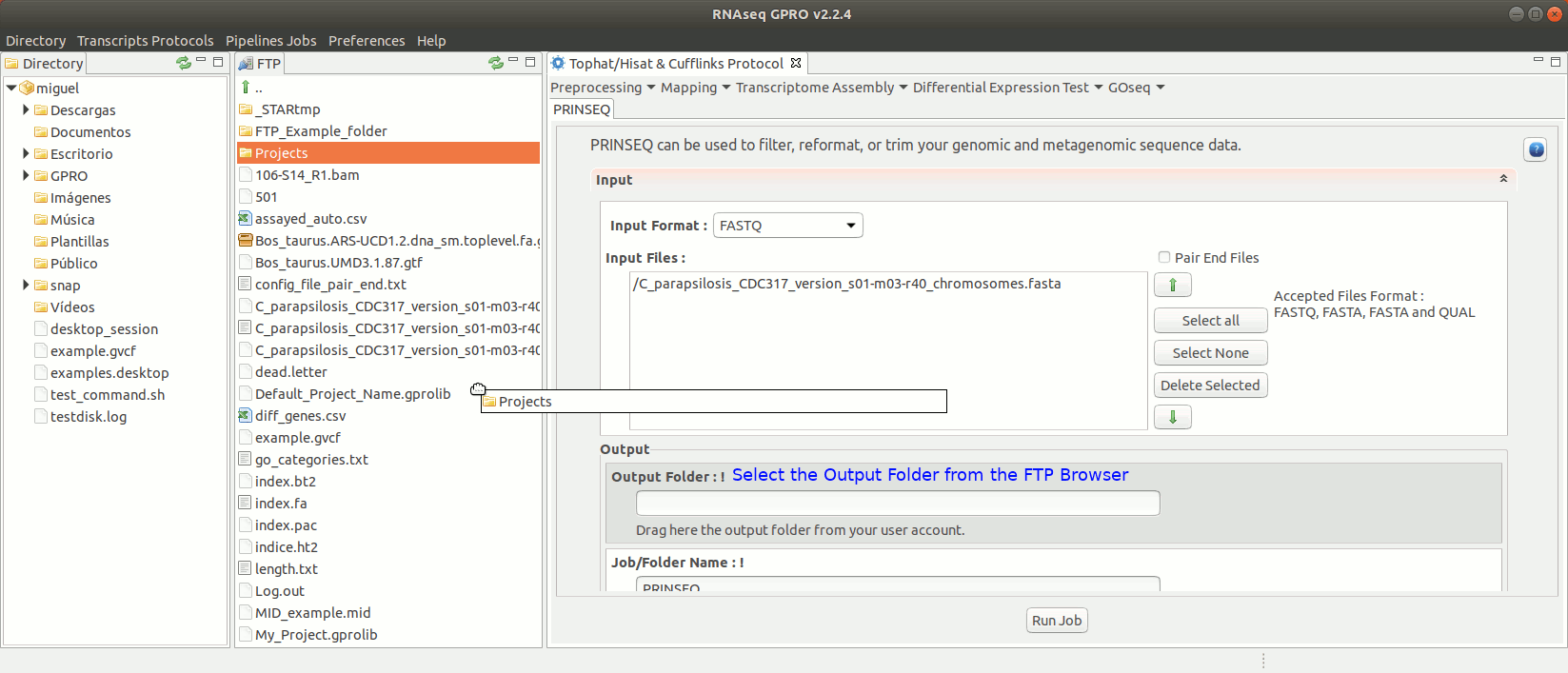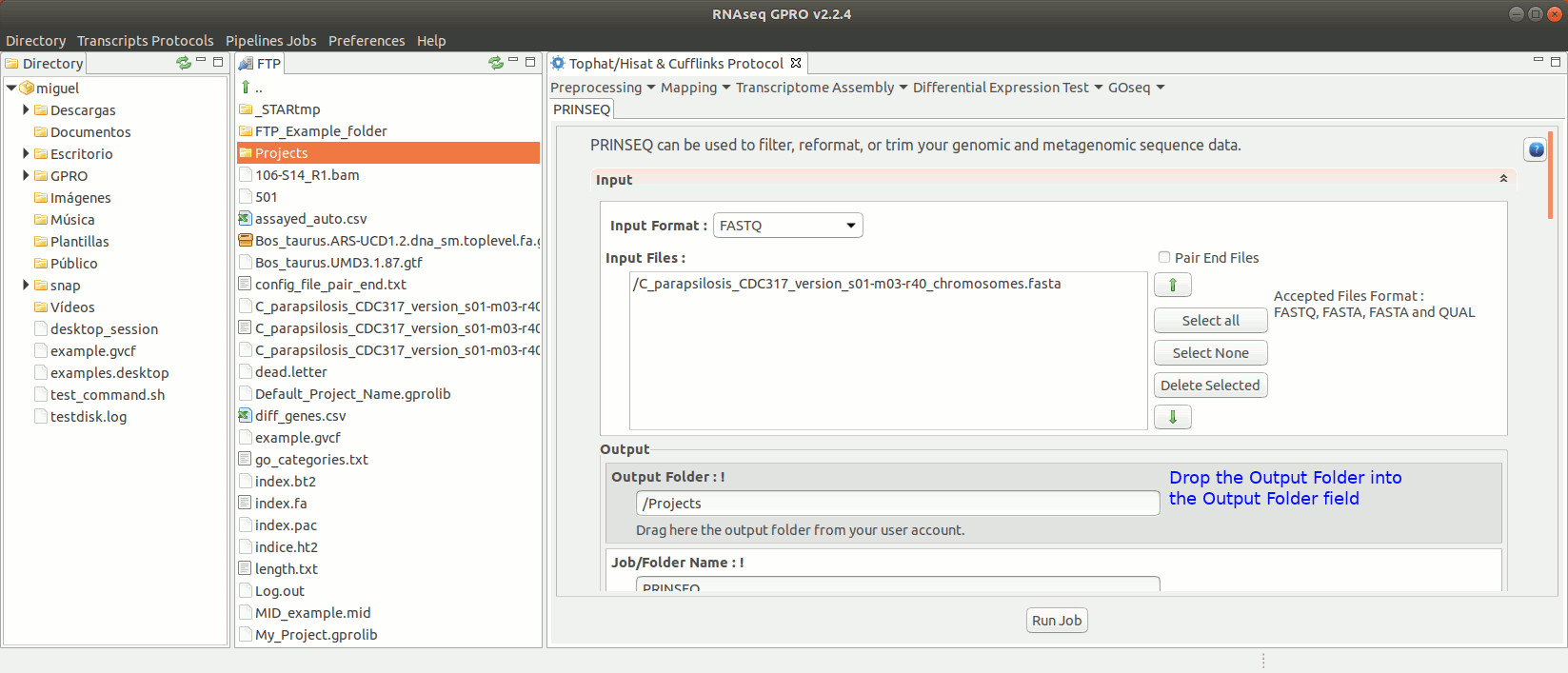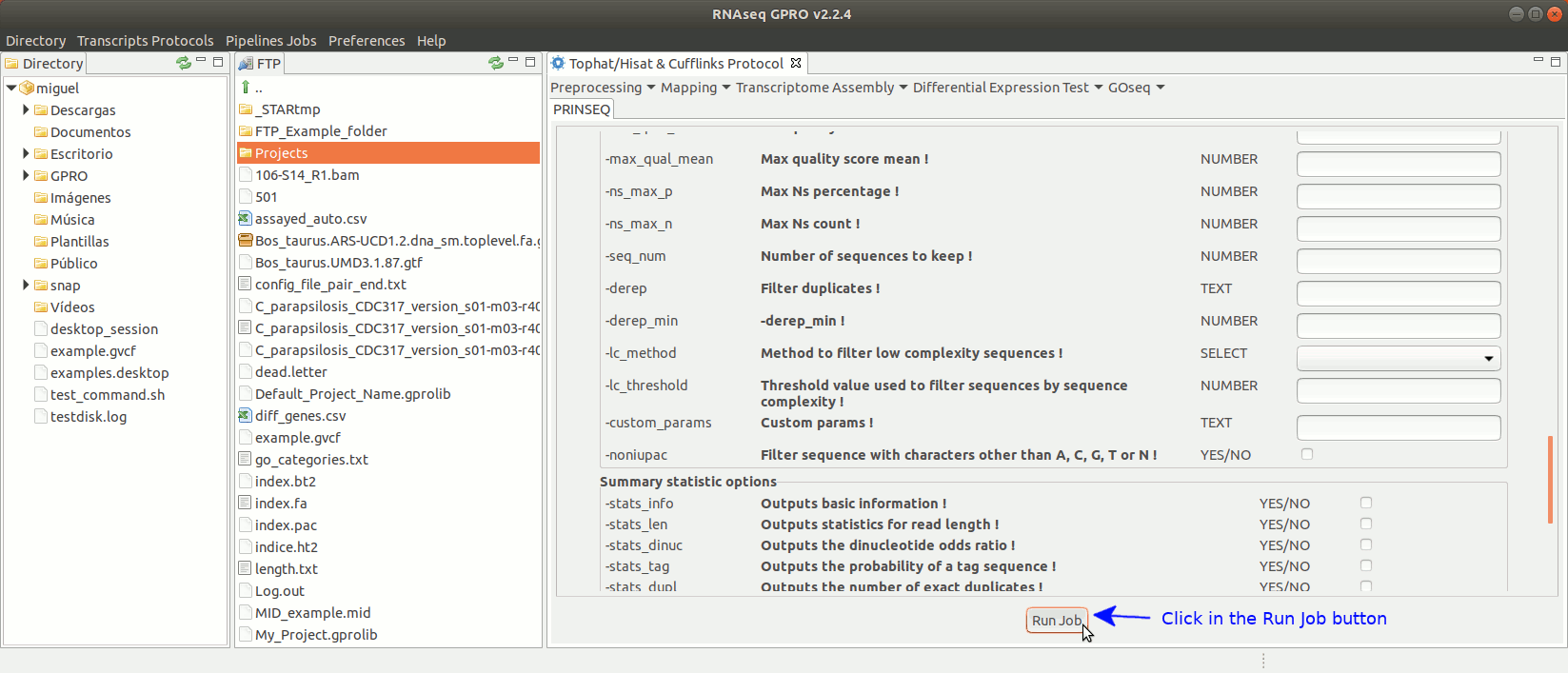
Figure 10: Using the GPRO interface for Prinseq.
- Trimming & Cleaning: Trimmomatic
Trimmomatic (Bolger et al. 2014) is trimming tool specific for paired-end and single-end reads obtained via Illumina’s NGS technology that can perform a variety of trimming tasks. For more information see the trimmomatic manual at "http://www.usadellab.org/cms/?page=trimmomatic".
To run Trimmomatic go to [“Preprocessing→Trimming & Cleaning → trimmomatic”] and follow Fig. 11.
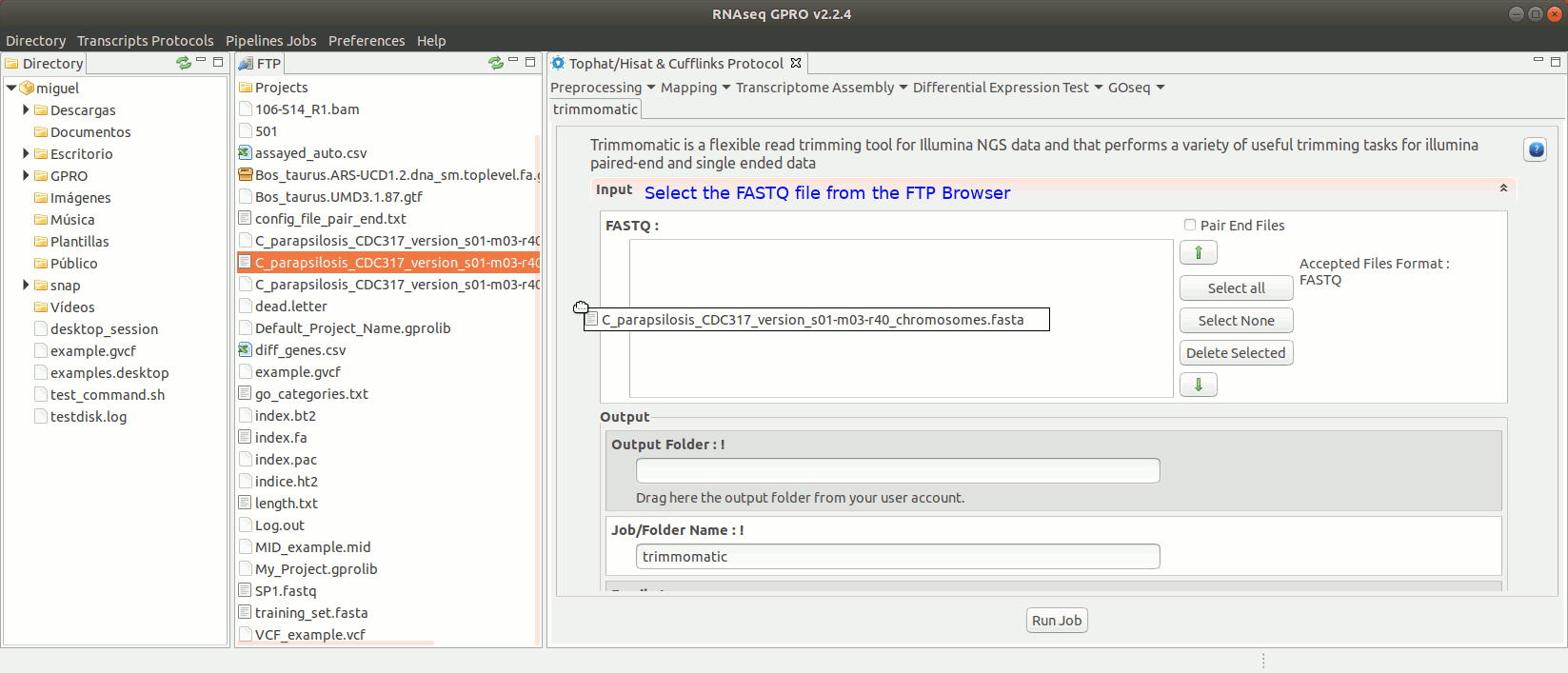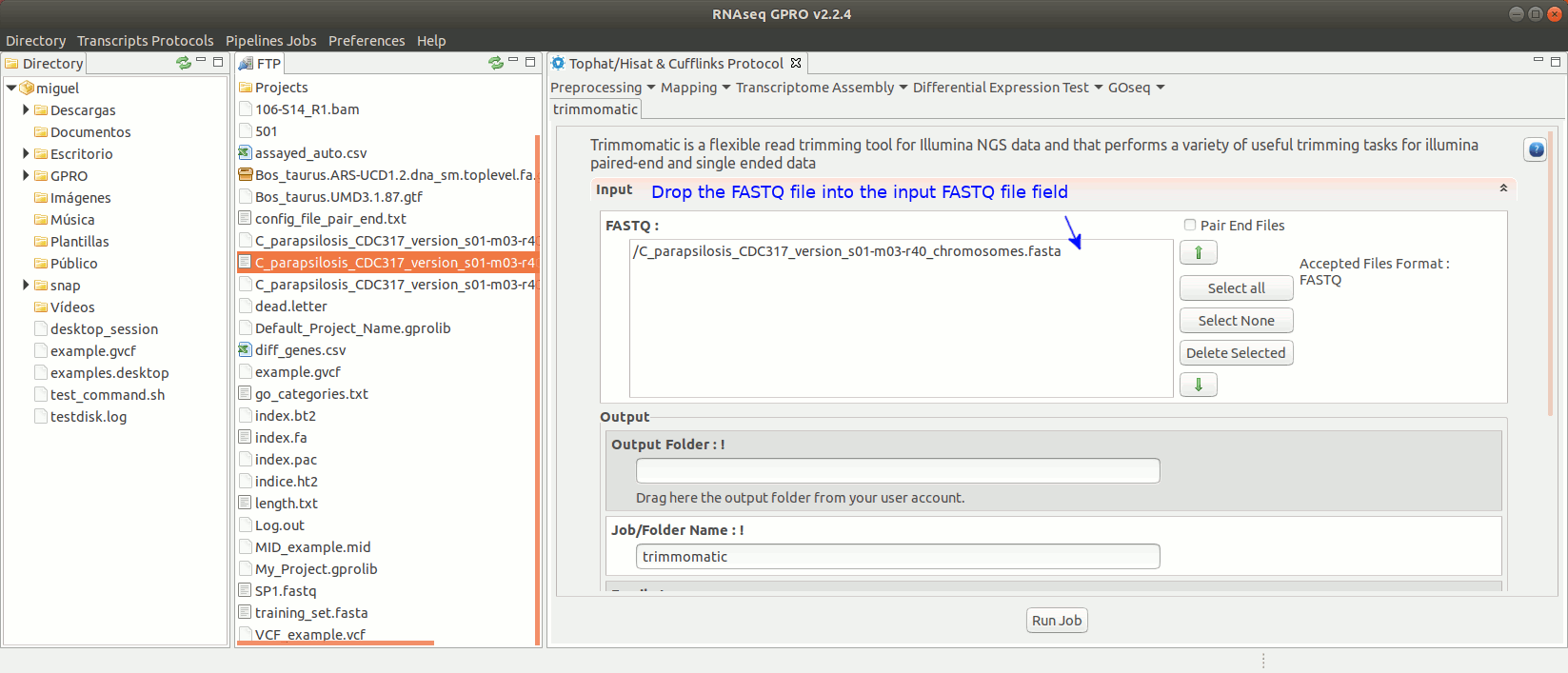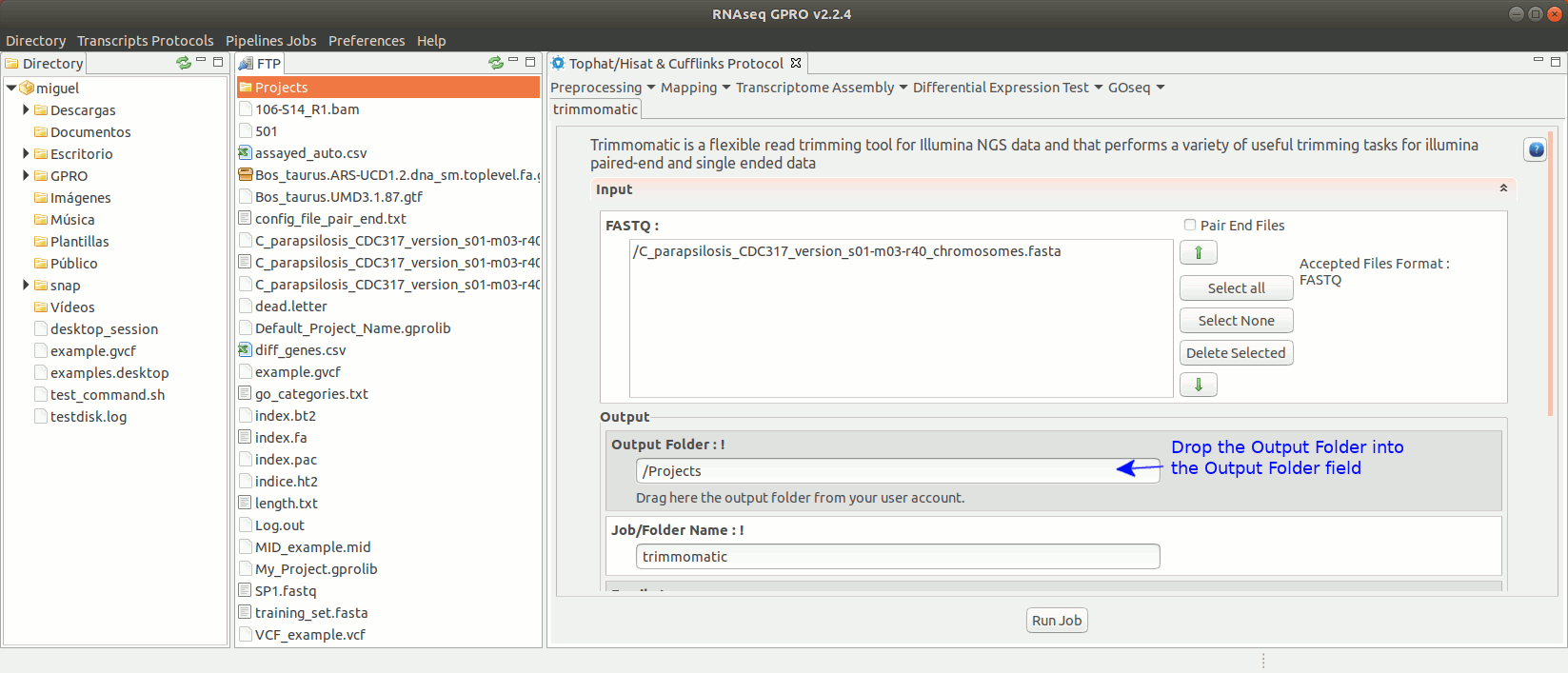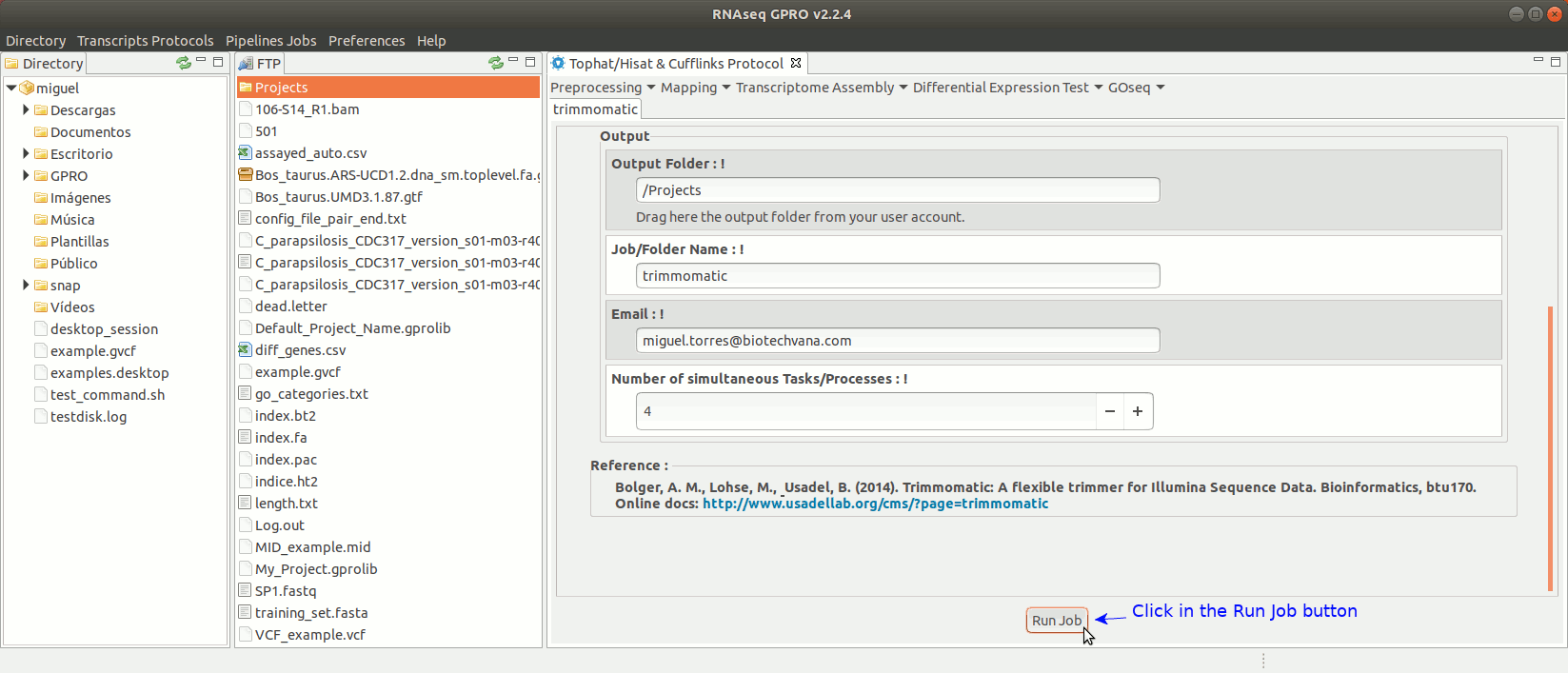
Figure 11: Using the GPRO interface for Trimmomatic.
- Trimming & Cleaning: FastxToolKit
FASTX-Toolkit (Hannon Lab 2016) is a collection of tools for the preprocessing of Fasta/Fastq files that include the following:
To run any of the FastxToolKit go to [“Preprocessing→ Trimming & Cleaning → Fastx-Toolkit”] and follow Fig. 12.
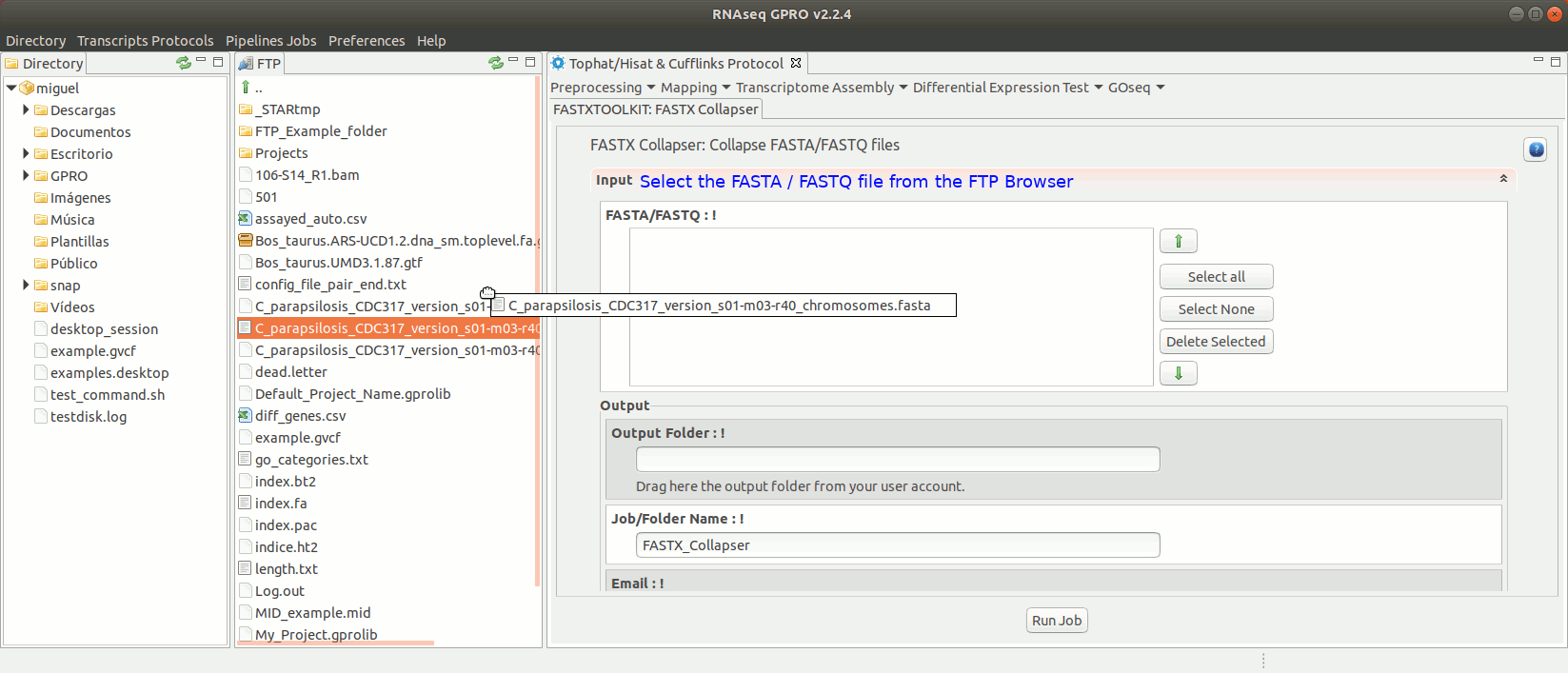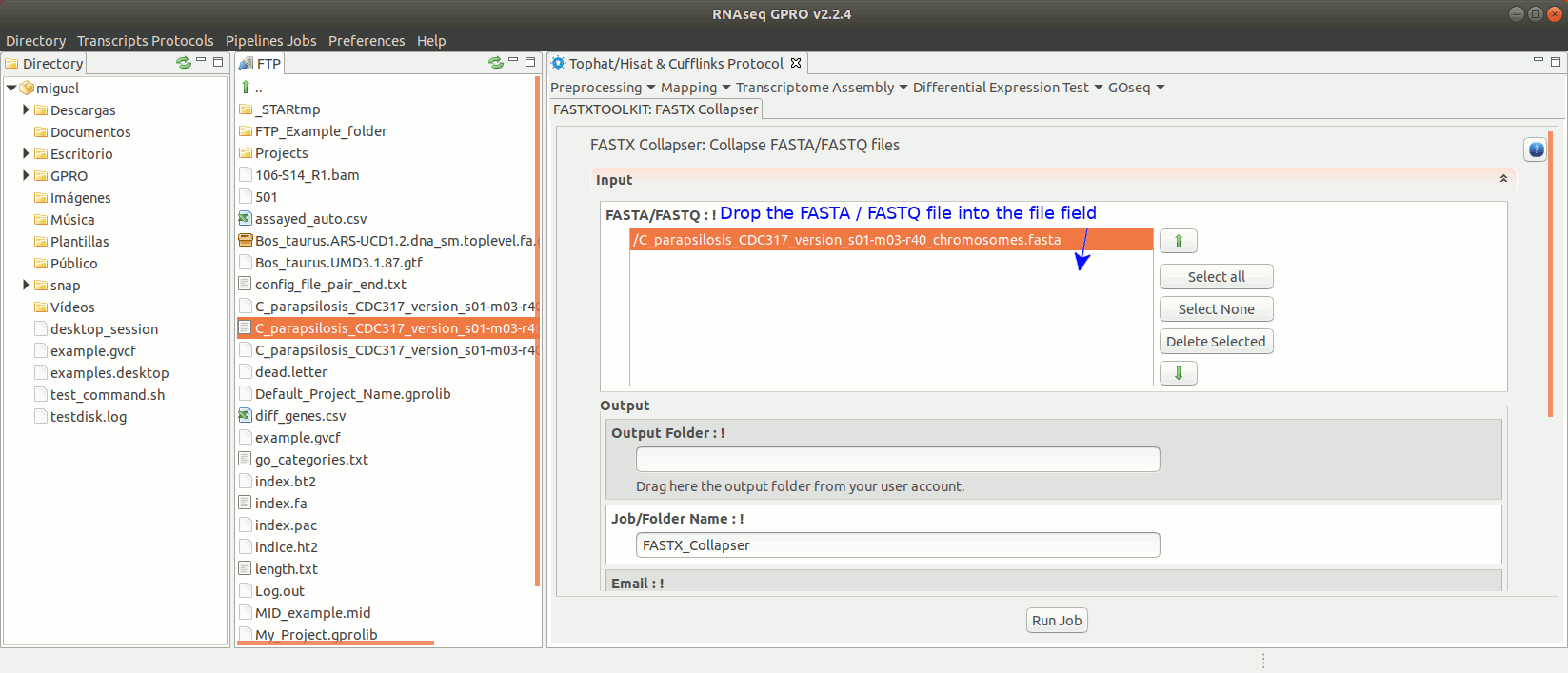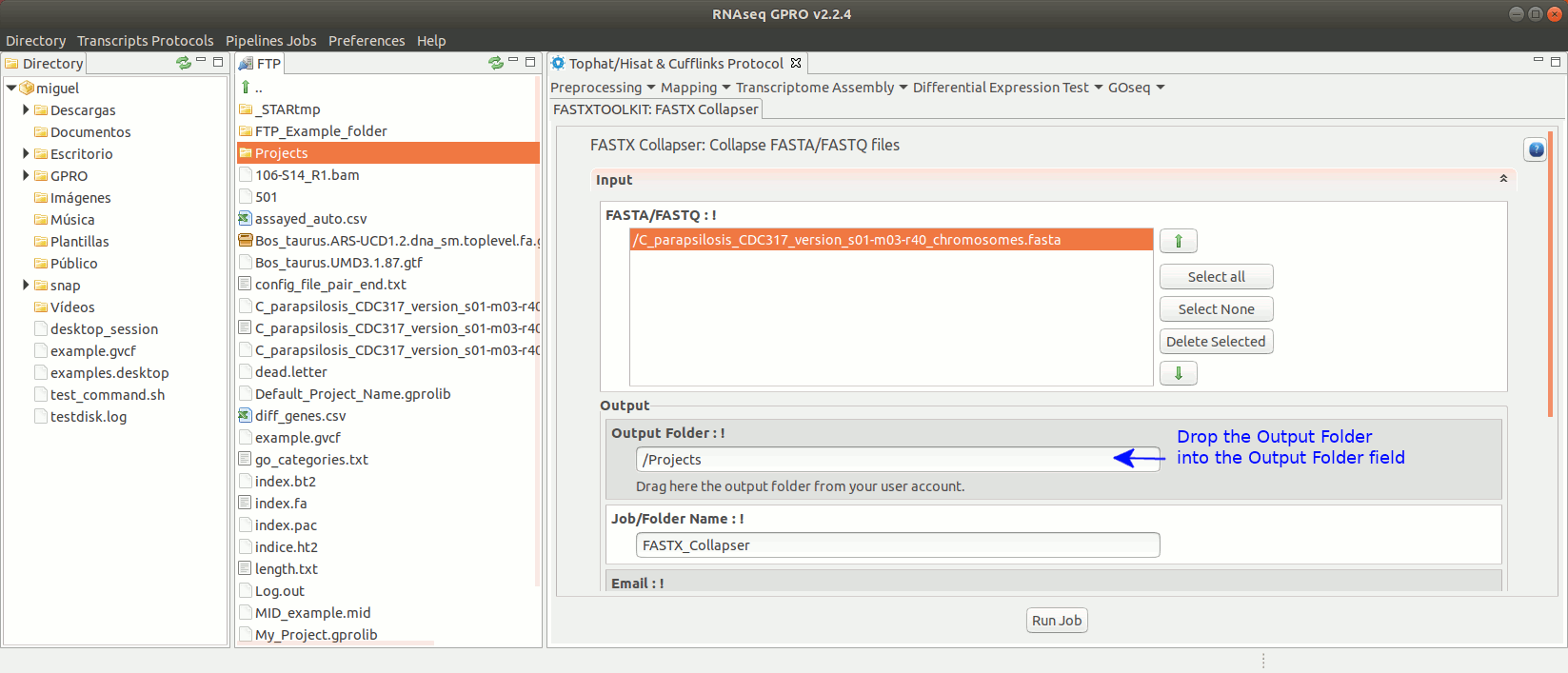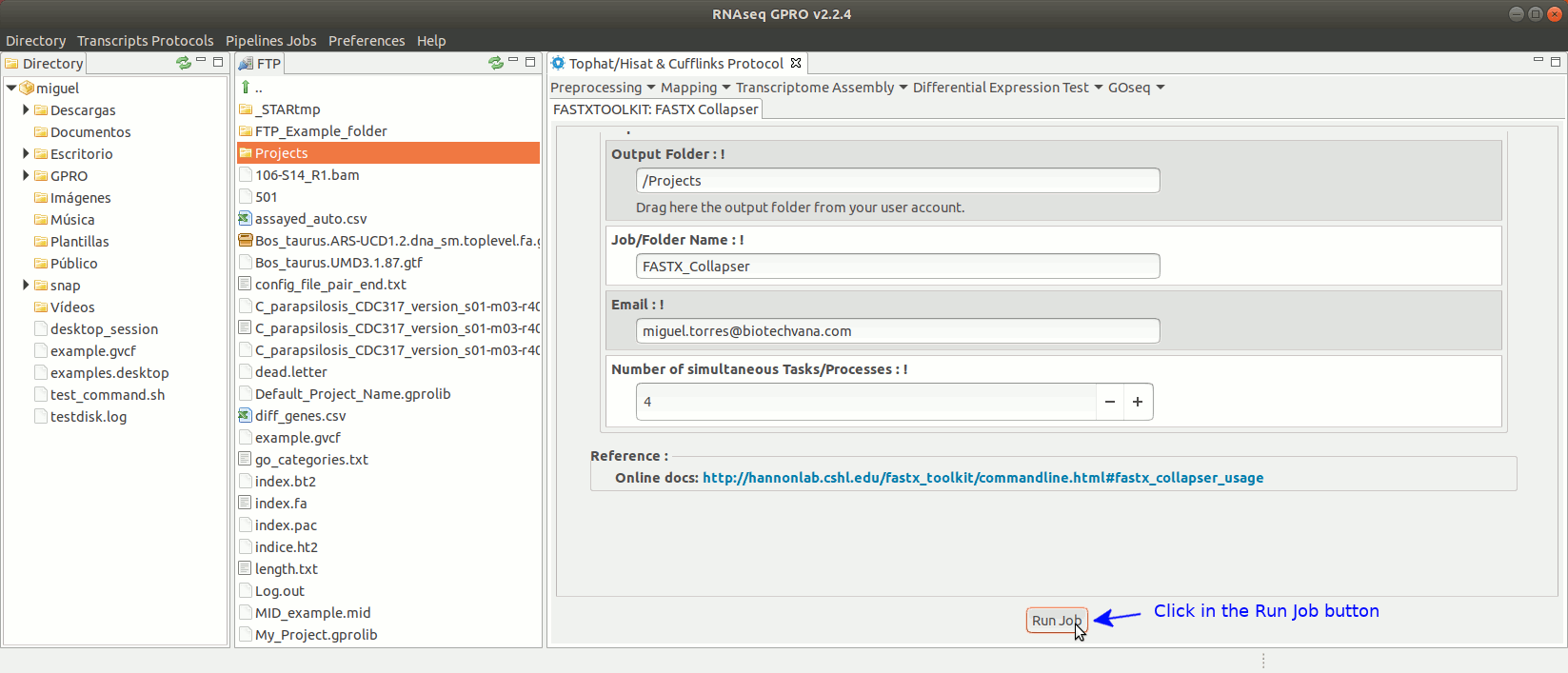
Figure 12: Example of GPRO interface for a tool (FASTX collapser) of Fastx-Toolkit.
- PrepSeq: FastqCollapser
FastqCollapser is used to remove duplicate reads from fastq files based on their sequence content.
To run FastqCollapser go to[ Preprocessing→ PrepSeq → FastqCollapser ] and proceed as shown in Fig. 13.
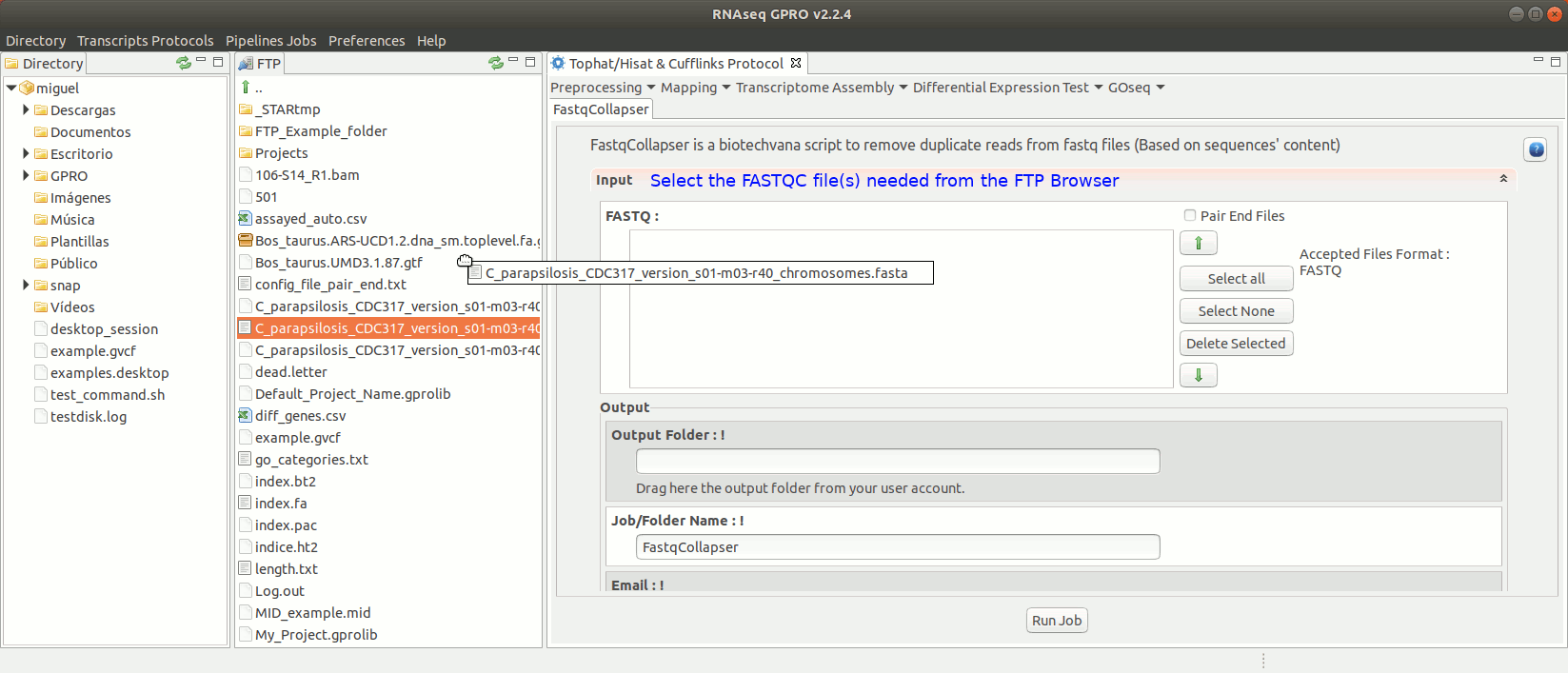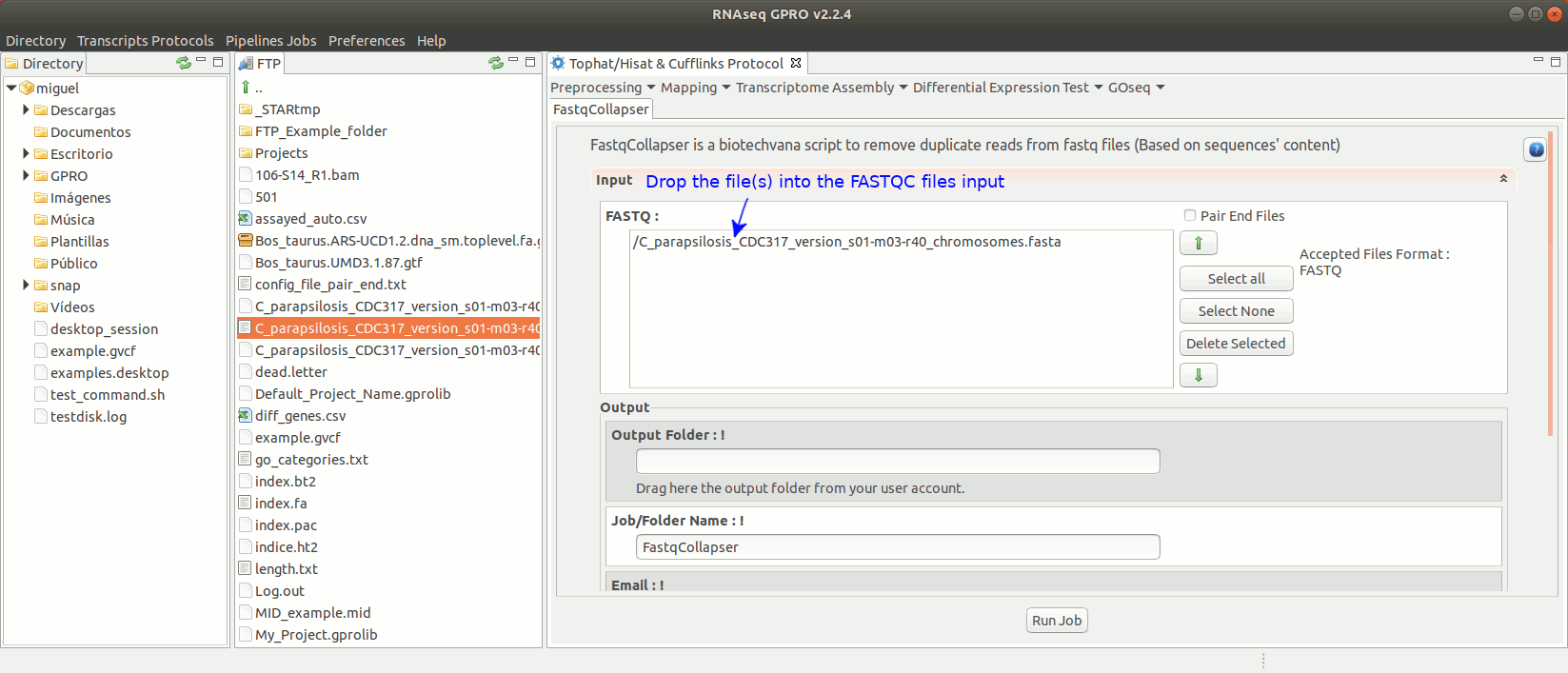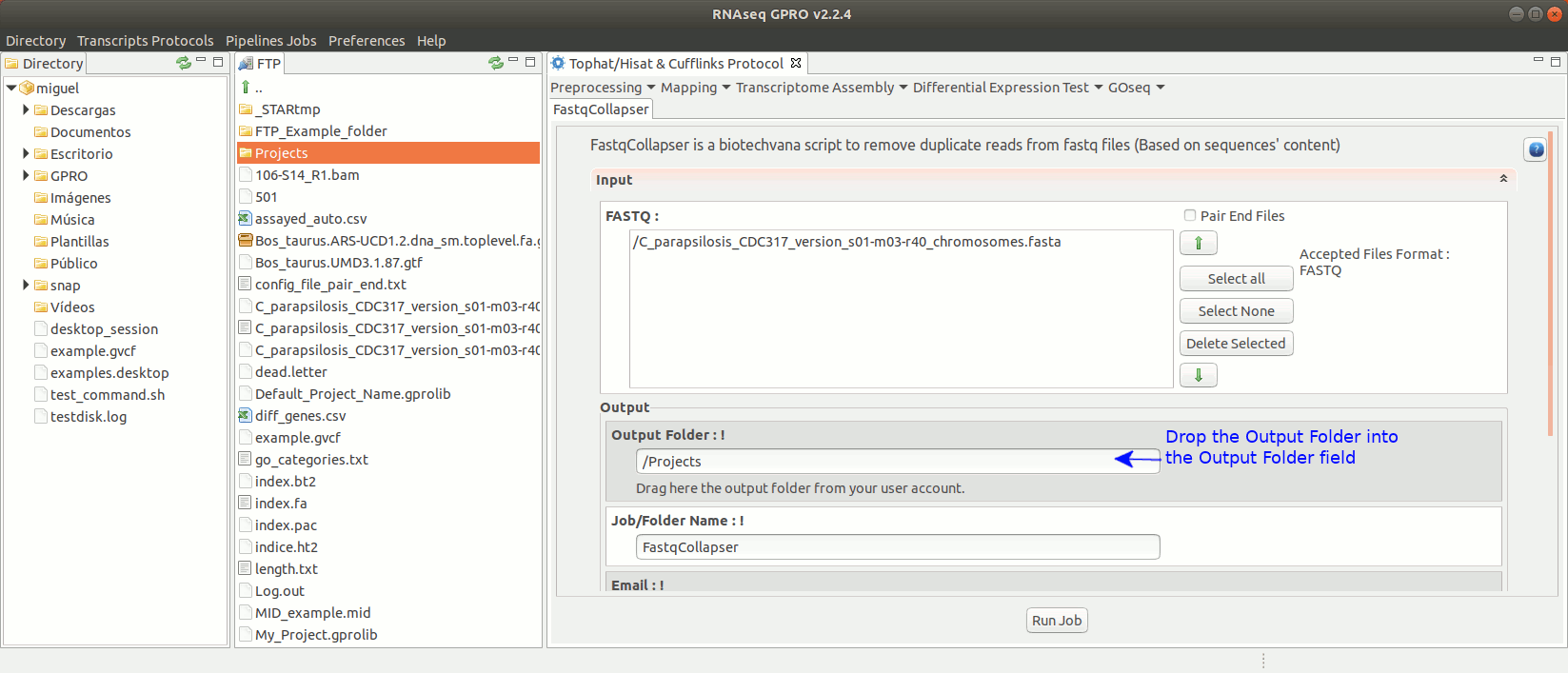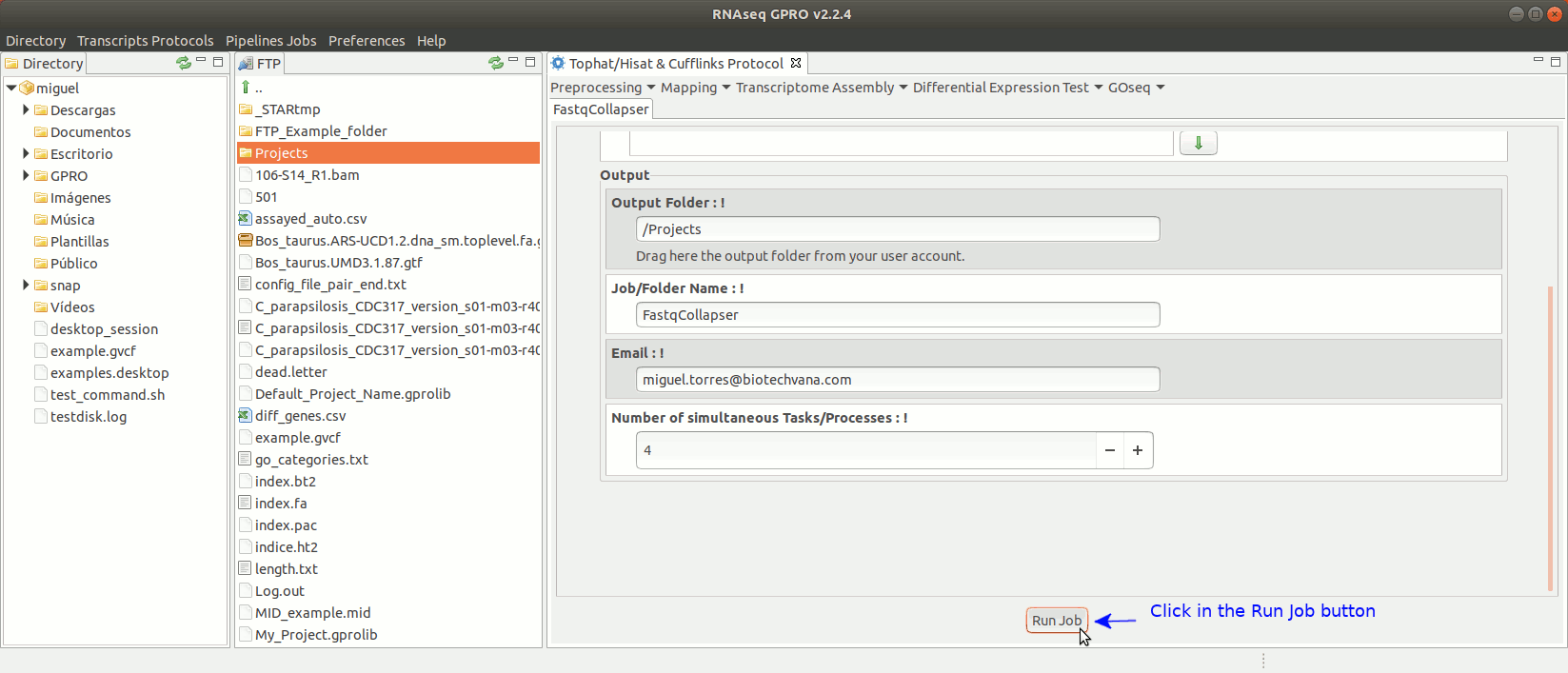
Figure 13: Using the GPRO interface for FastQCollapser.
- Trimming & Cleaning: FastqIntersect
FastqIntersect is a script that compares the information of two pair-end files that have been independently preprocessed and the information on both files to edit them keeping only those reads, and in the same order, that are present in both files (mate reads). This tool used when the number of reads obtained does not match the output of the execution of any preprocessing tool in each file individually the other. This is because assembly/mapping processes require that the files match in the number and the sort of reads. Please note that both Prinseq and Trimmomatic already have a function to intersect reads by ticking the ‘pair end files’ box. Thus, FastqIntersect will only need to be run in either those cases where the ’pair end files’ box has not been selected. FastqIntersect will also not need to be used when Cutadapt has been used, since this tool does not implement intersecting functions.
To run FastqIntersect go to [ Preprocessing→ PrepSeq → FastqIntersect ] and follow Fig. 14.
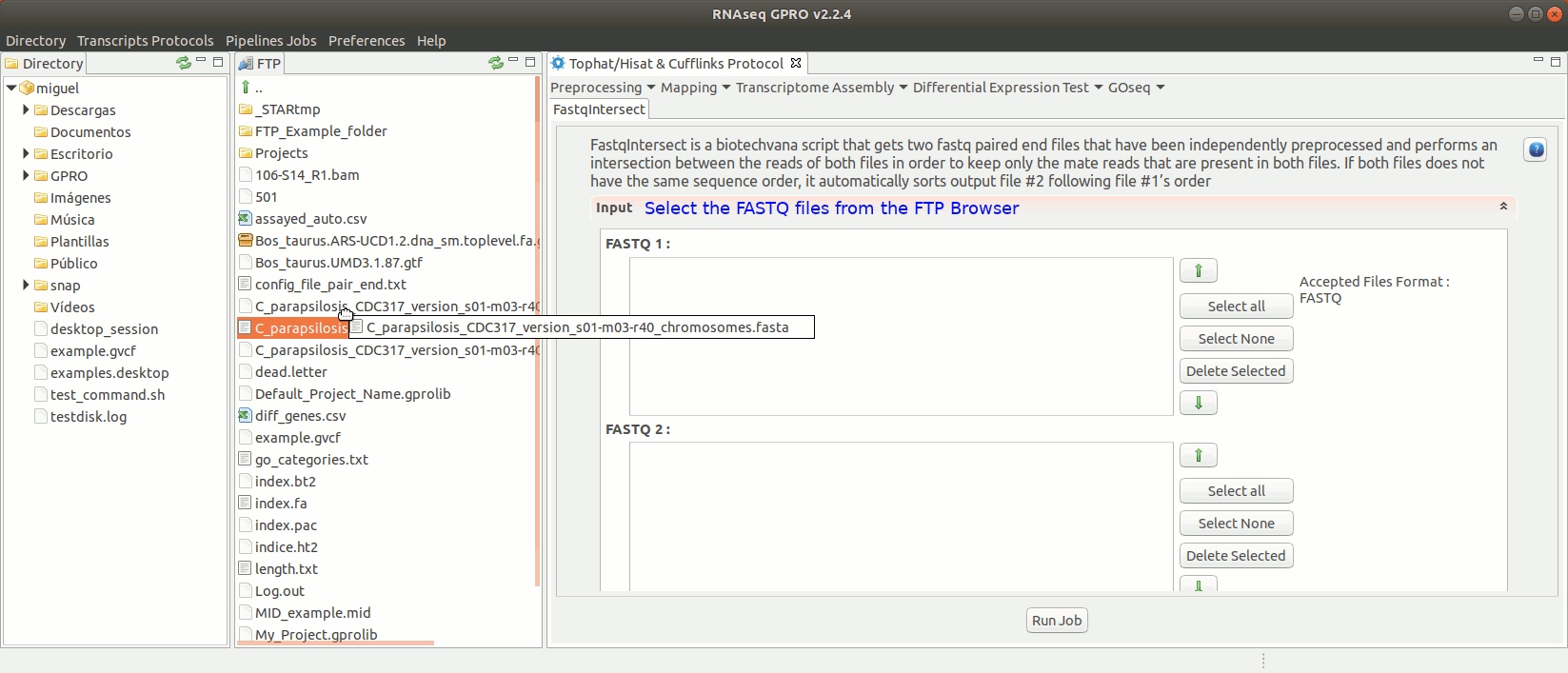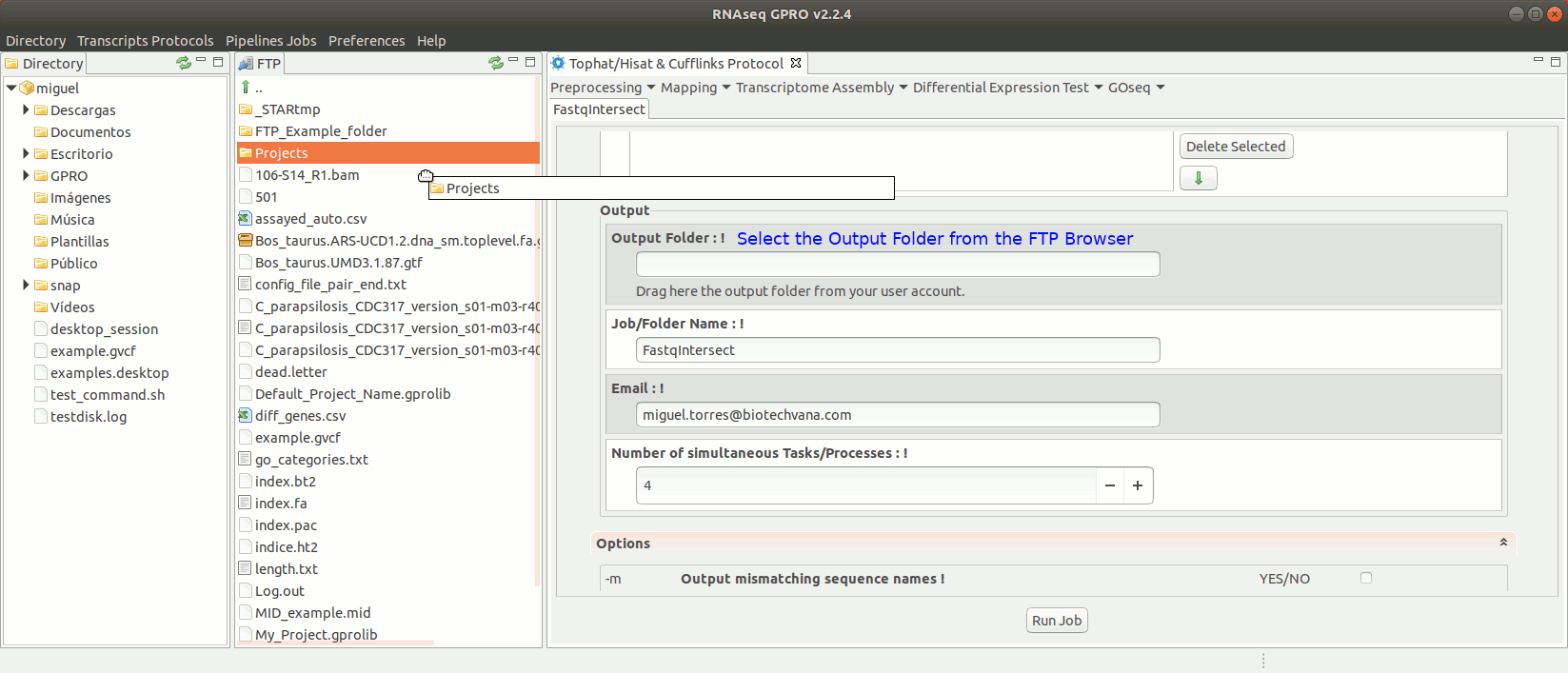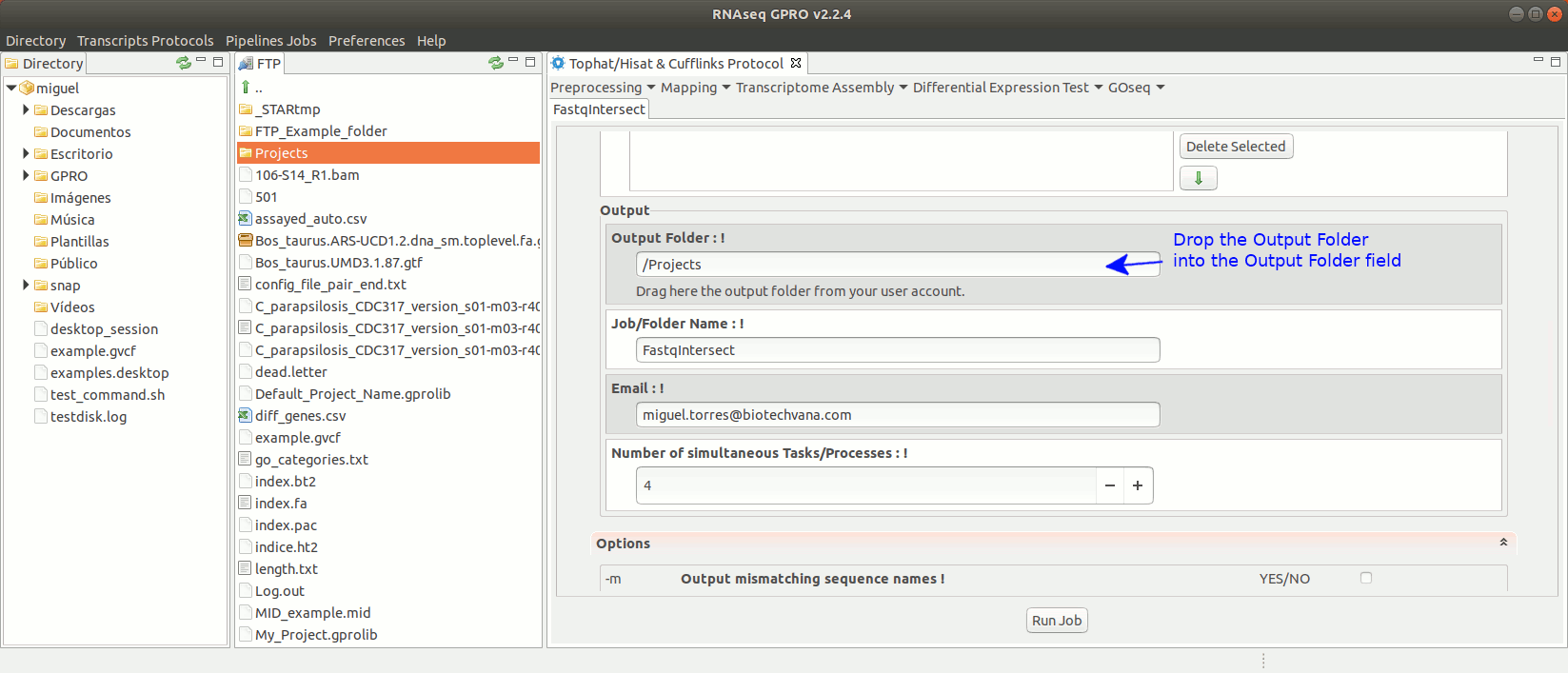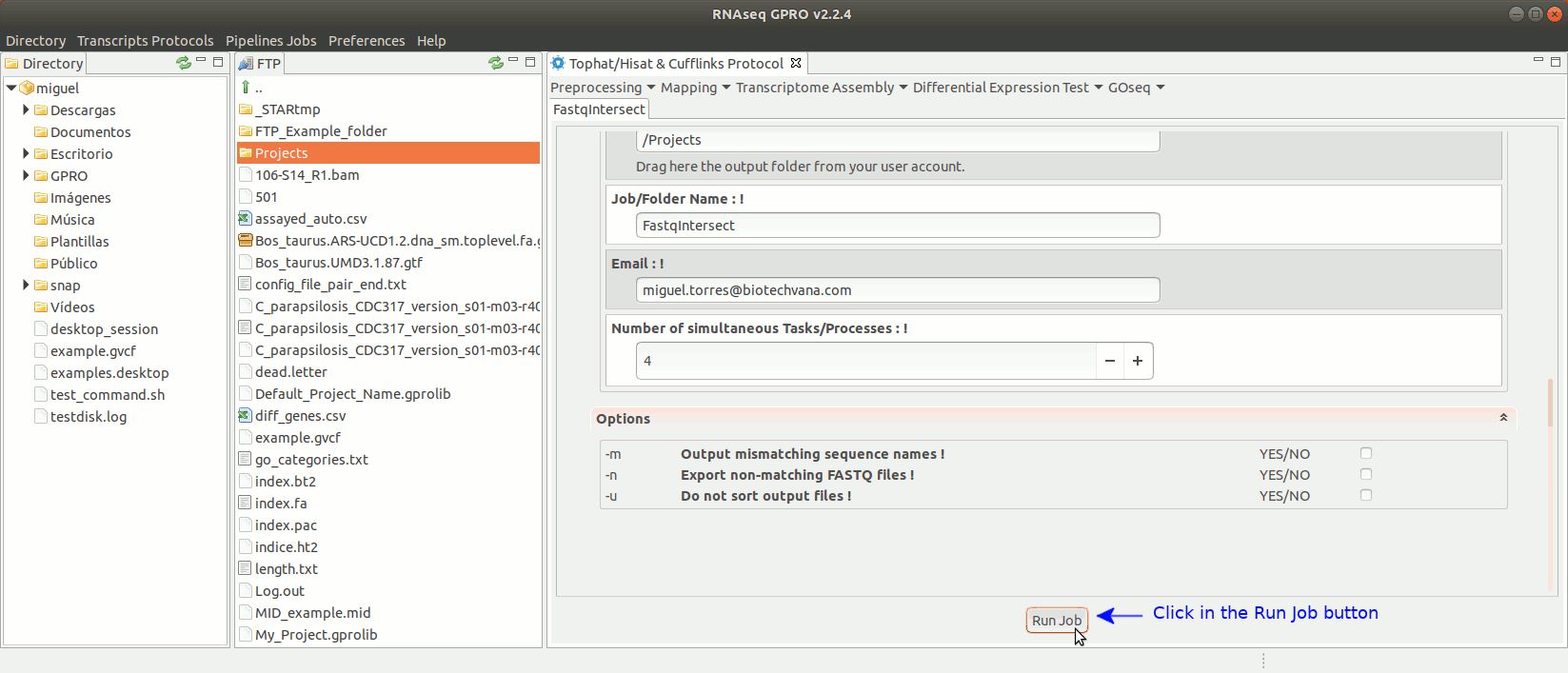
Figure 14: Using the GPRO interface fastqintersect.
- Mapping : Tophat
TopHat (Trapnell et al. 2012,Kim et al. 2013) ligns RNA-Seq reads to a reference genome identifying exon-exon splice junctions. For more information, see the Tophat manual at "https://daehwankimlab.github.io/hisat2/manual/".
To run Tophat go to[Mapping→ Tophat ] and follow Fig. 15.
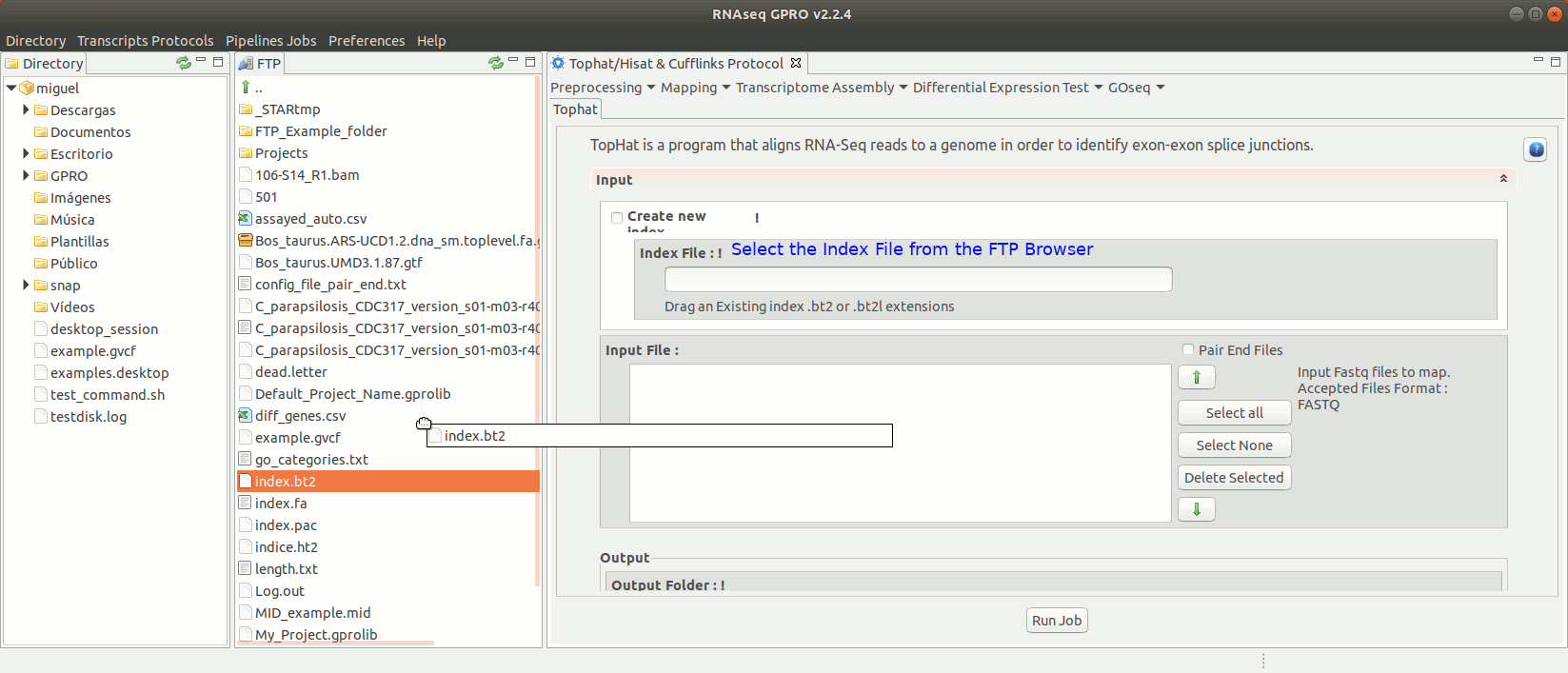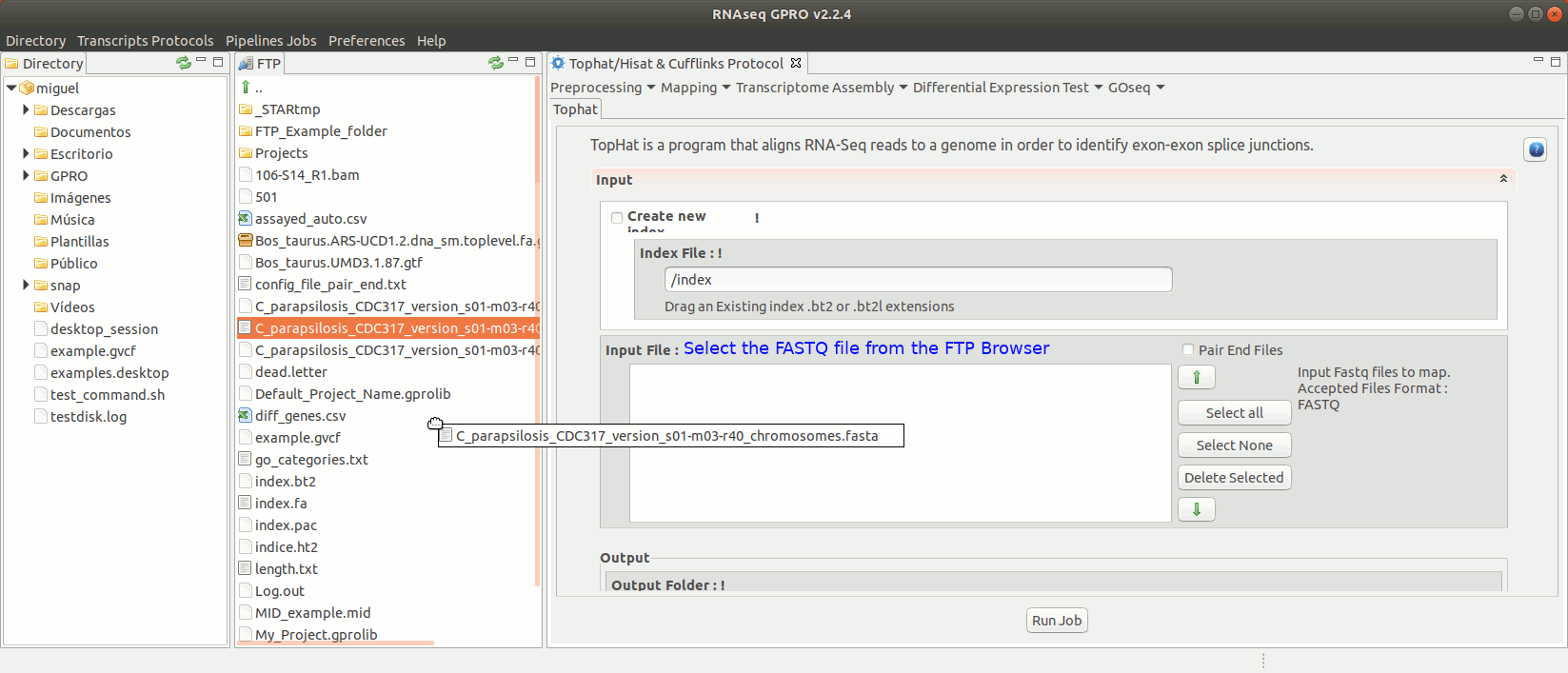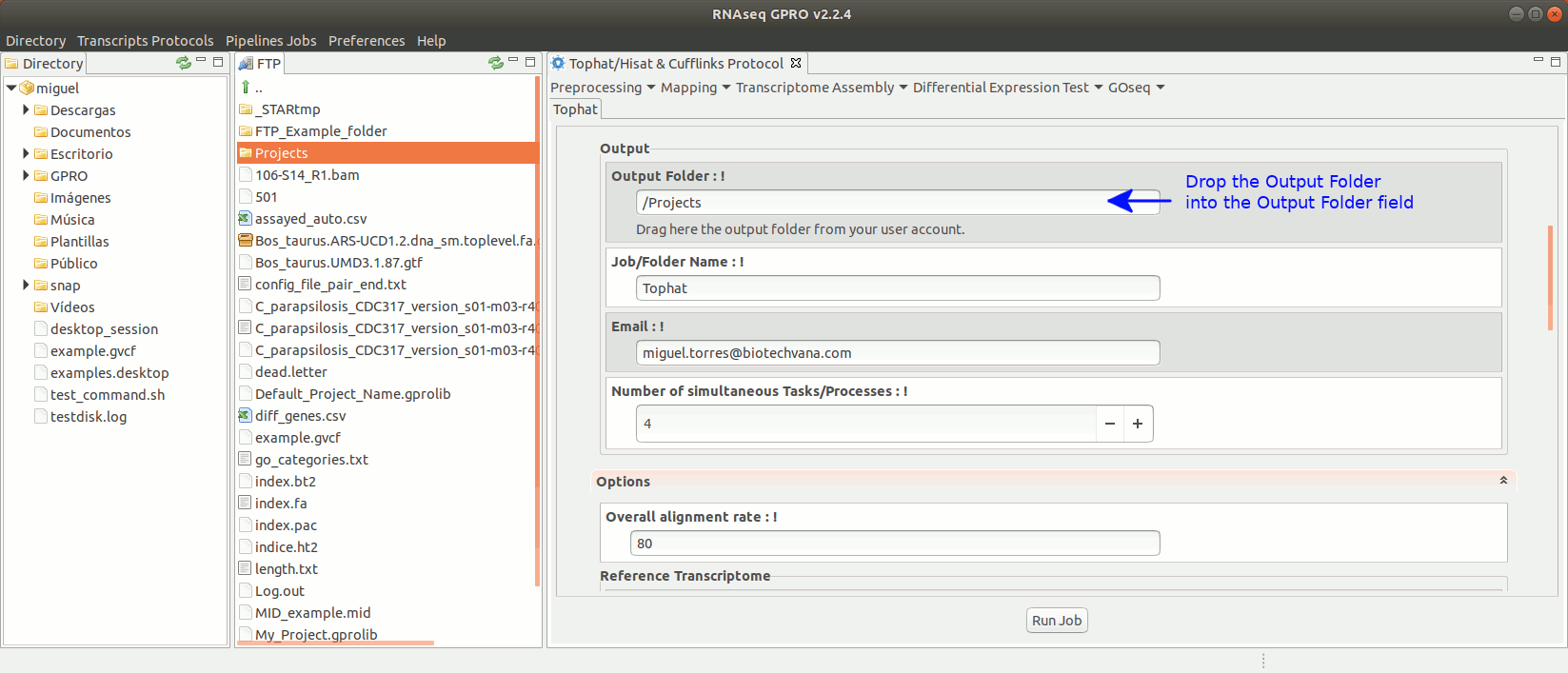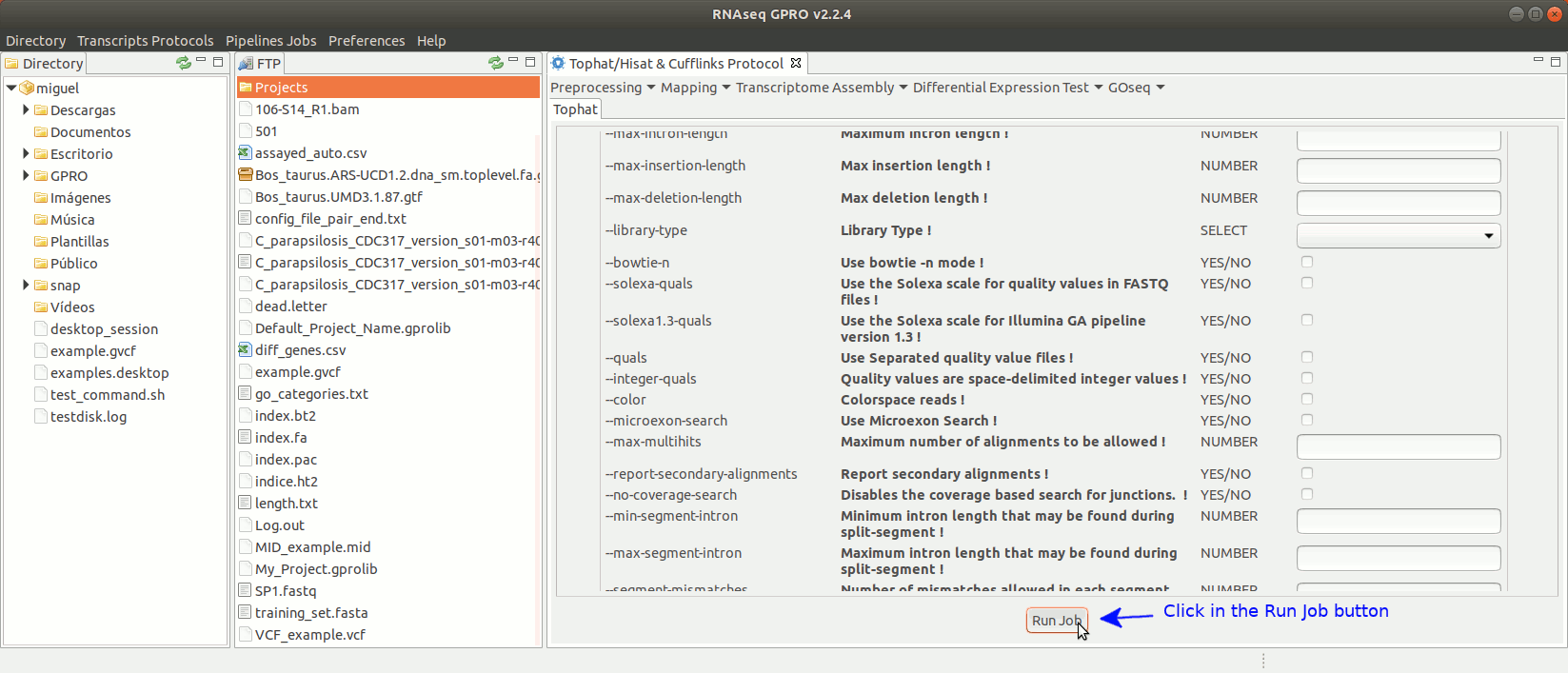
Figure 15: Using the GPRO interface for TopHat.
- Mapping : Hisat2
HISAT2 (Kim, Langmead, and Salzberg 2015) is a tool for mapping of sequencing reads (both DNA and RNA). For more information, see the HISAT2 manual at "https://ccb.jhu.edu/software/hisat2/manual.shtml" for more information.
To run Hisat2 go to [ Mapping→ Hisat2]and follow Fig. 16.
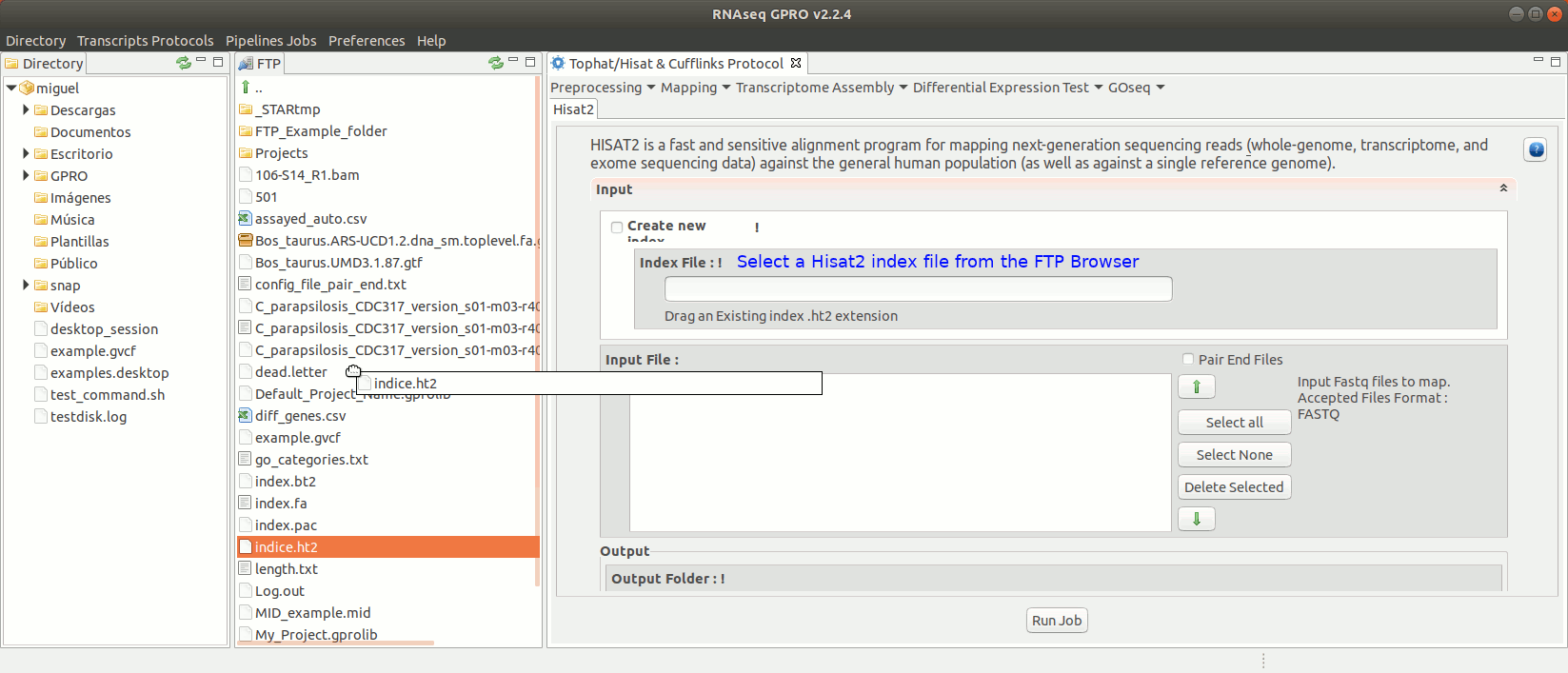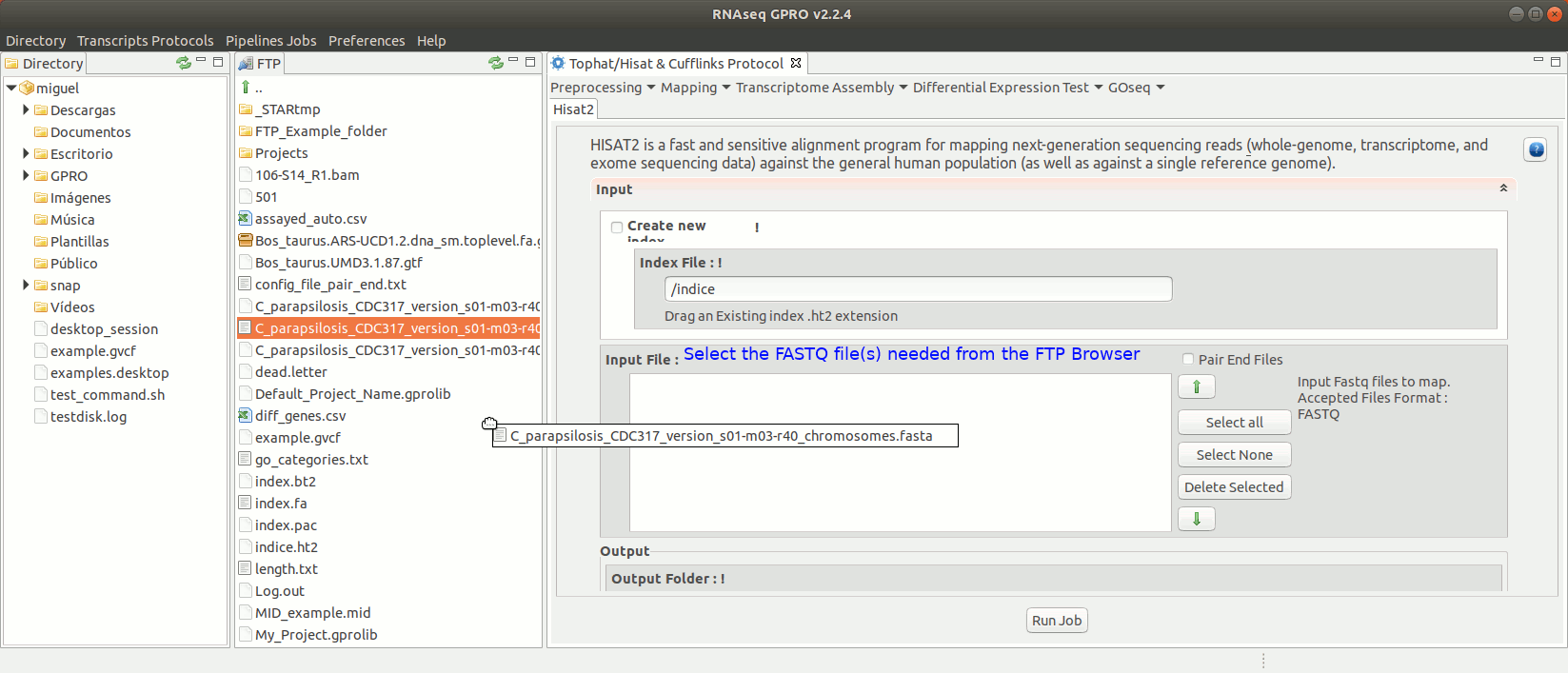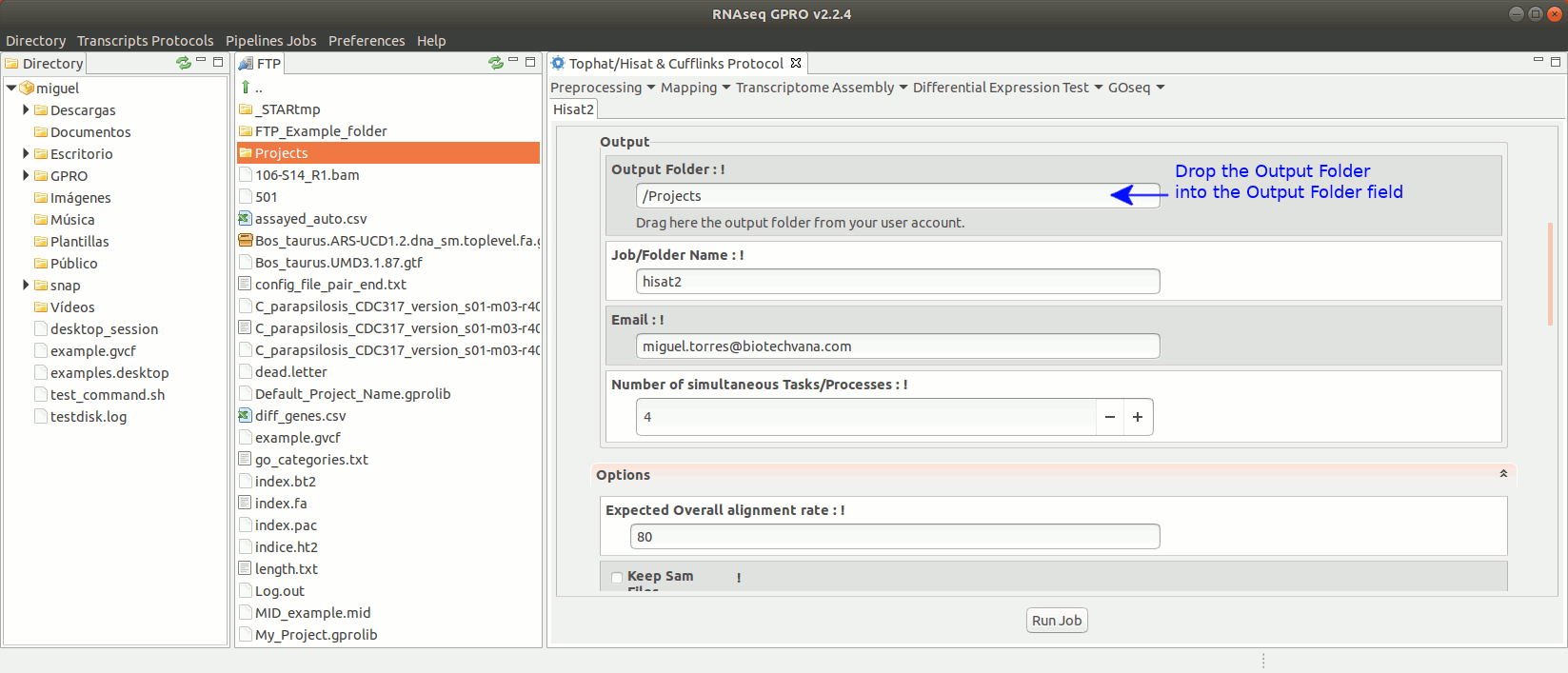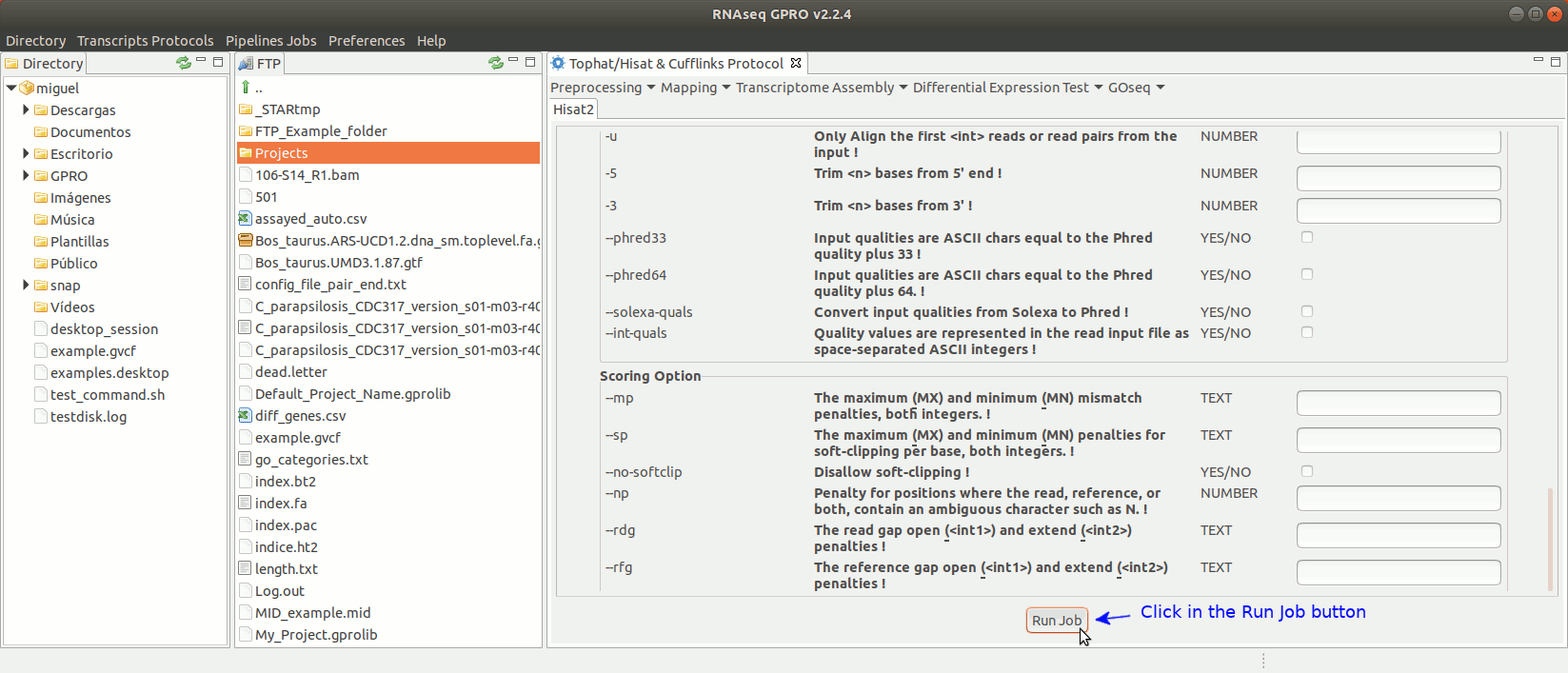
Figure 16: Using the GPRO interface for Hisat2.
- Transcriptome Assembly: Cufflinks
To run the Cufflinks protocol for transcriptome assembly go to[ Transcriptome Assembly→ Cufflinks ]and follow Fig. 17.
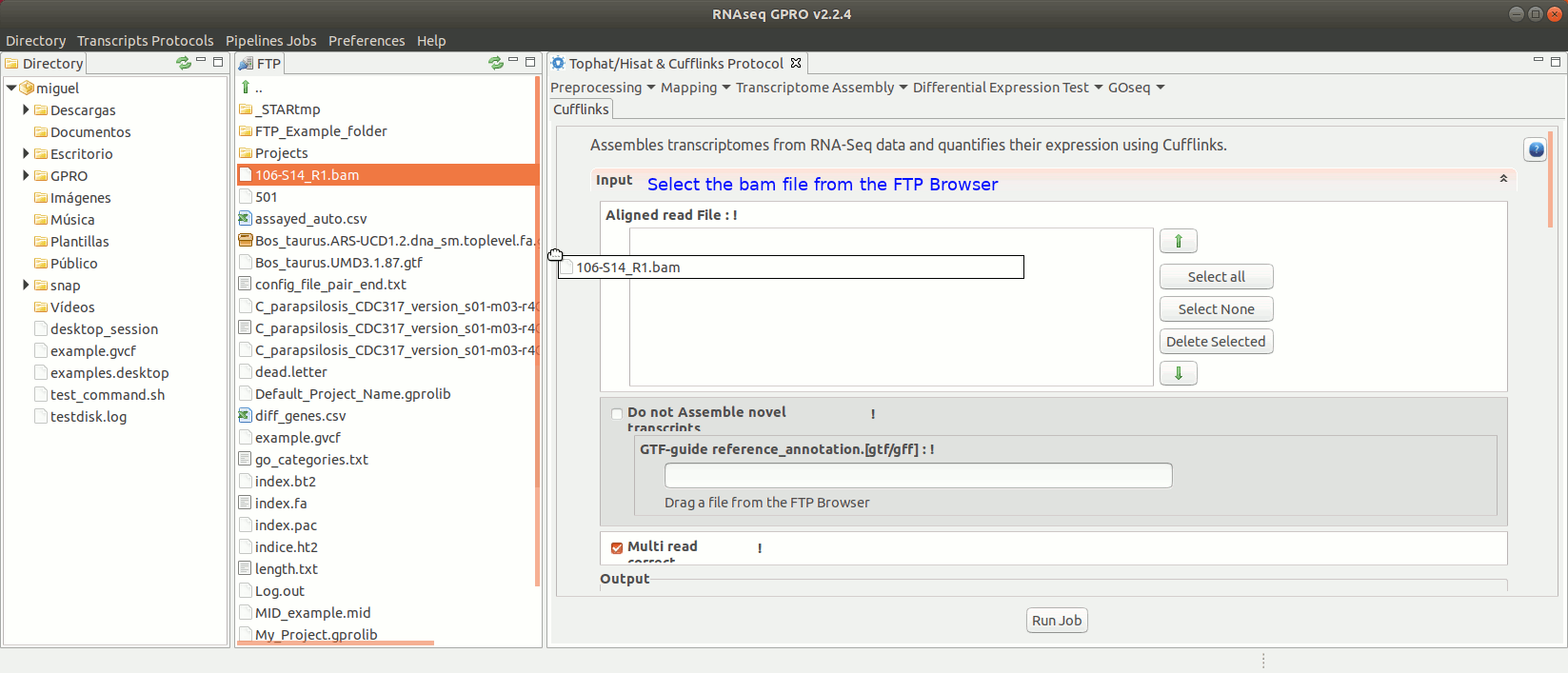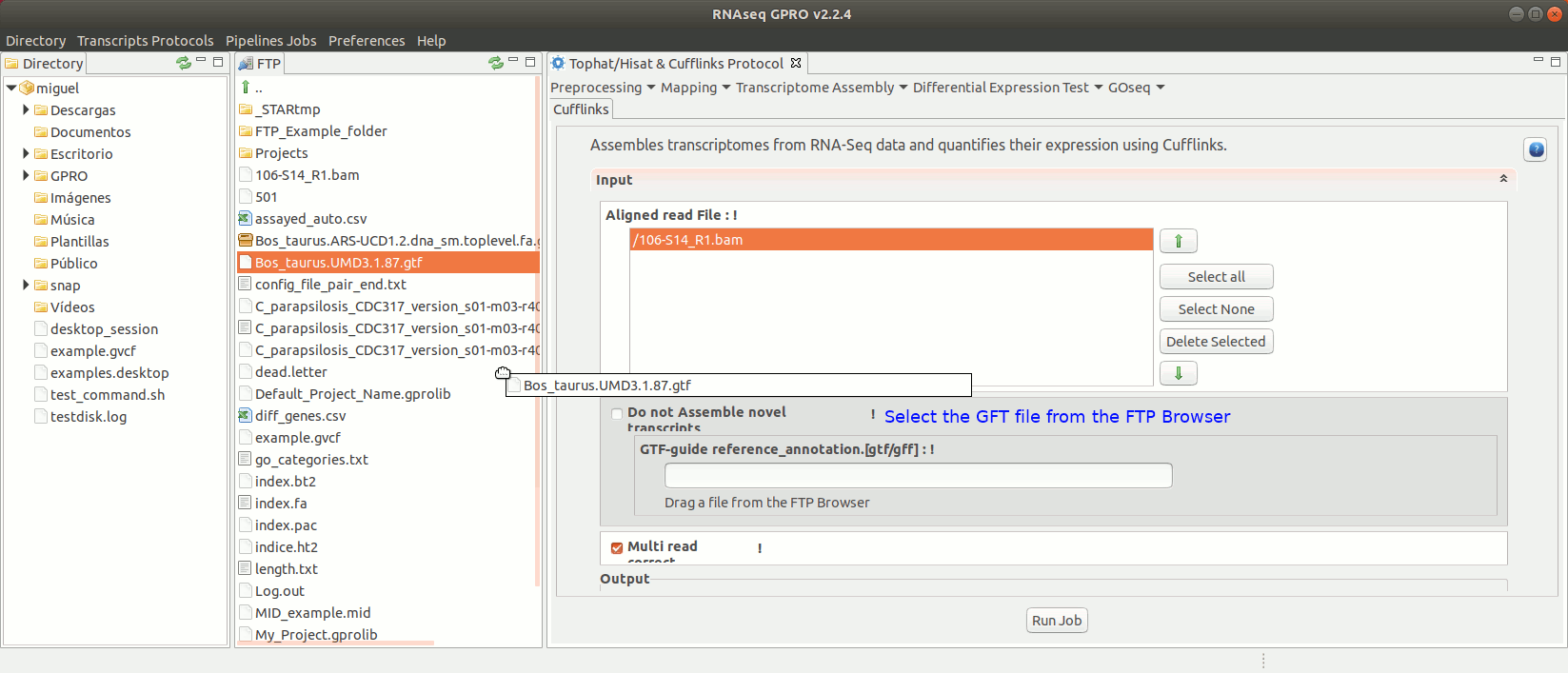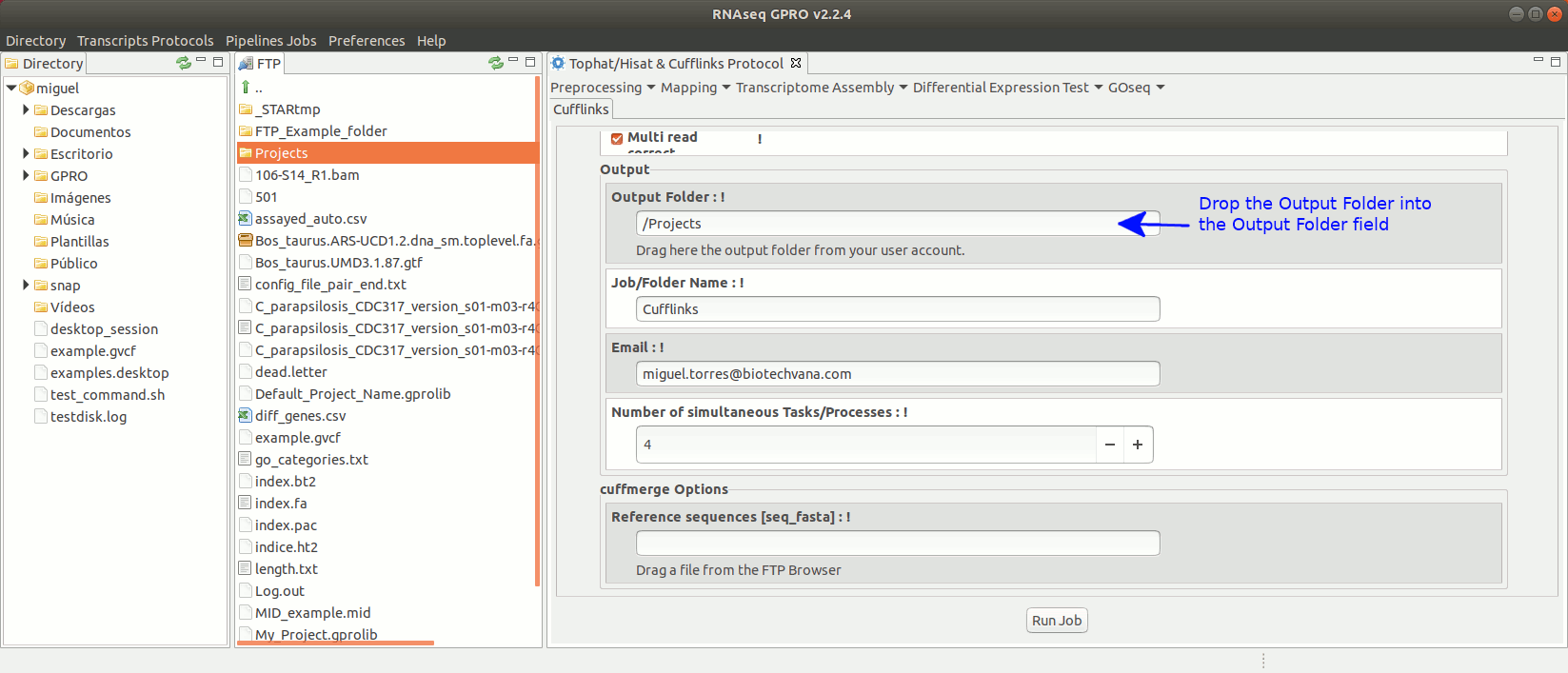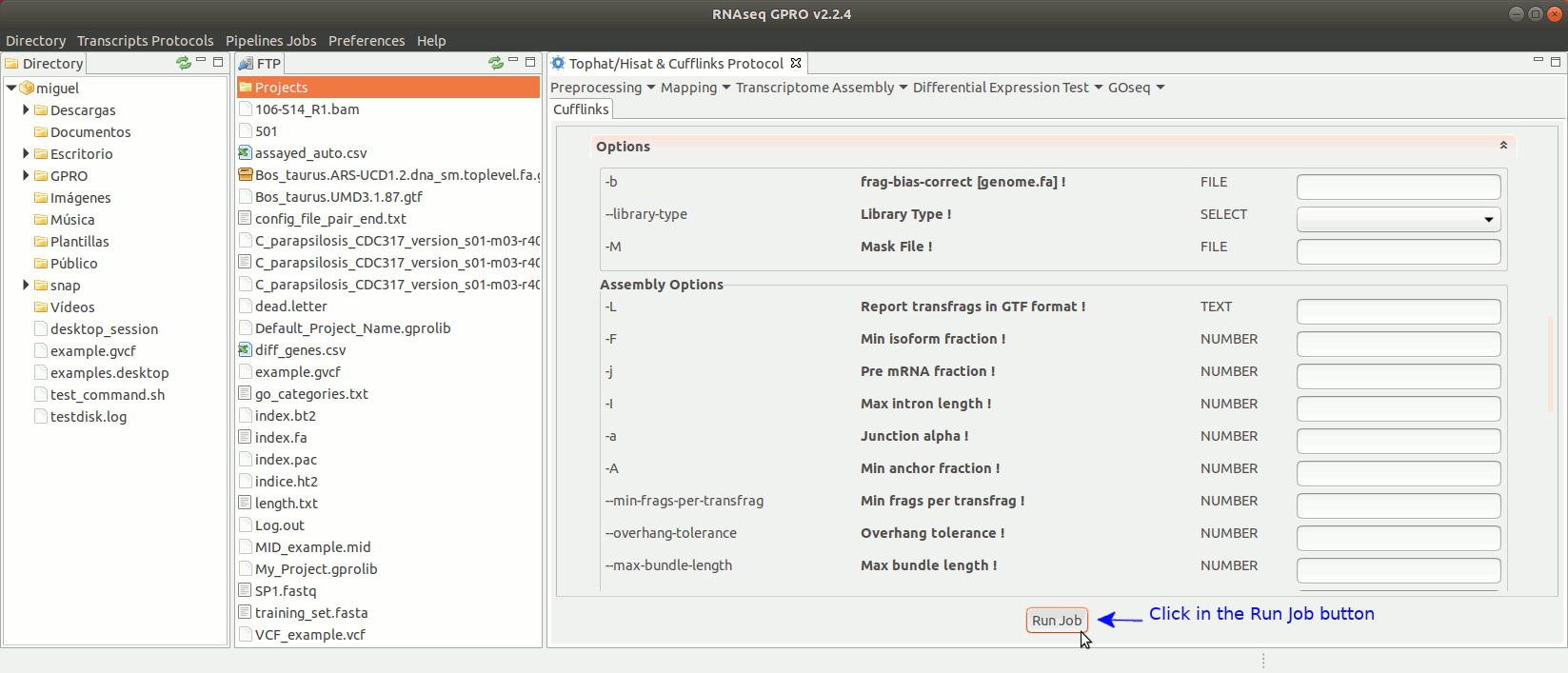
Figure 17: Using the GPRO interface for Cufflinks.
- Differential Expression Test: Cuffdiff
To run Cuffdiff go to [Diff Expression Test→ Cuffdiff] and follow Fig. 18.
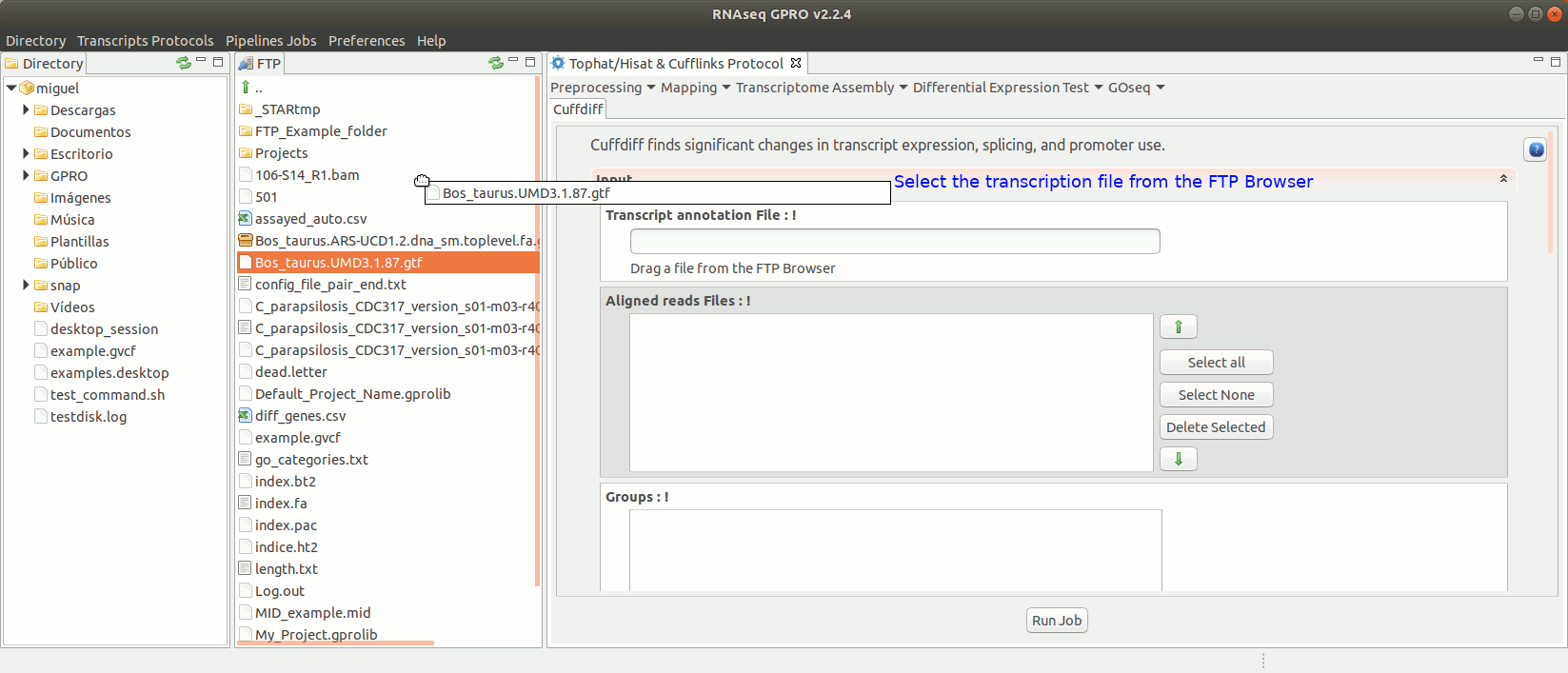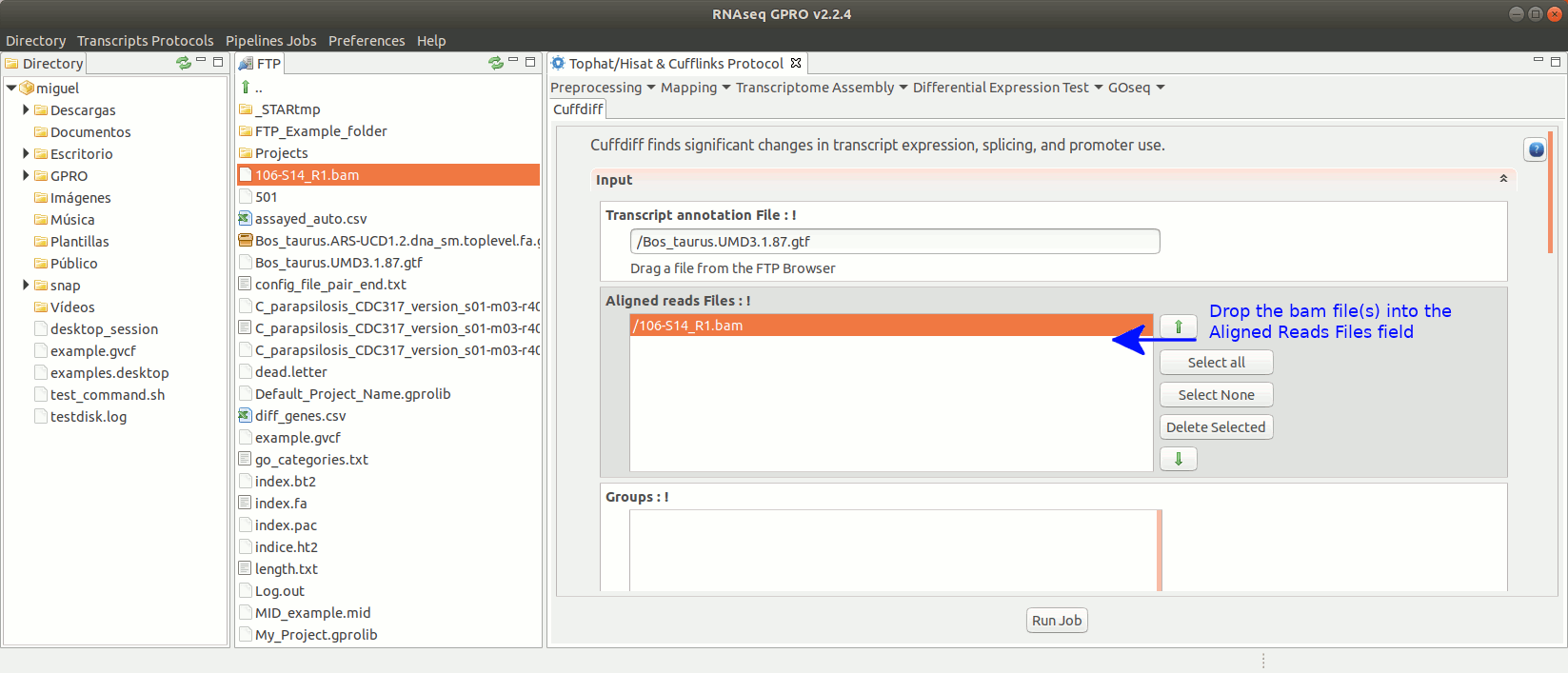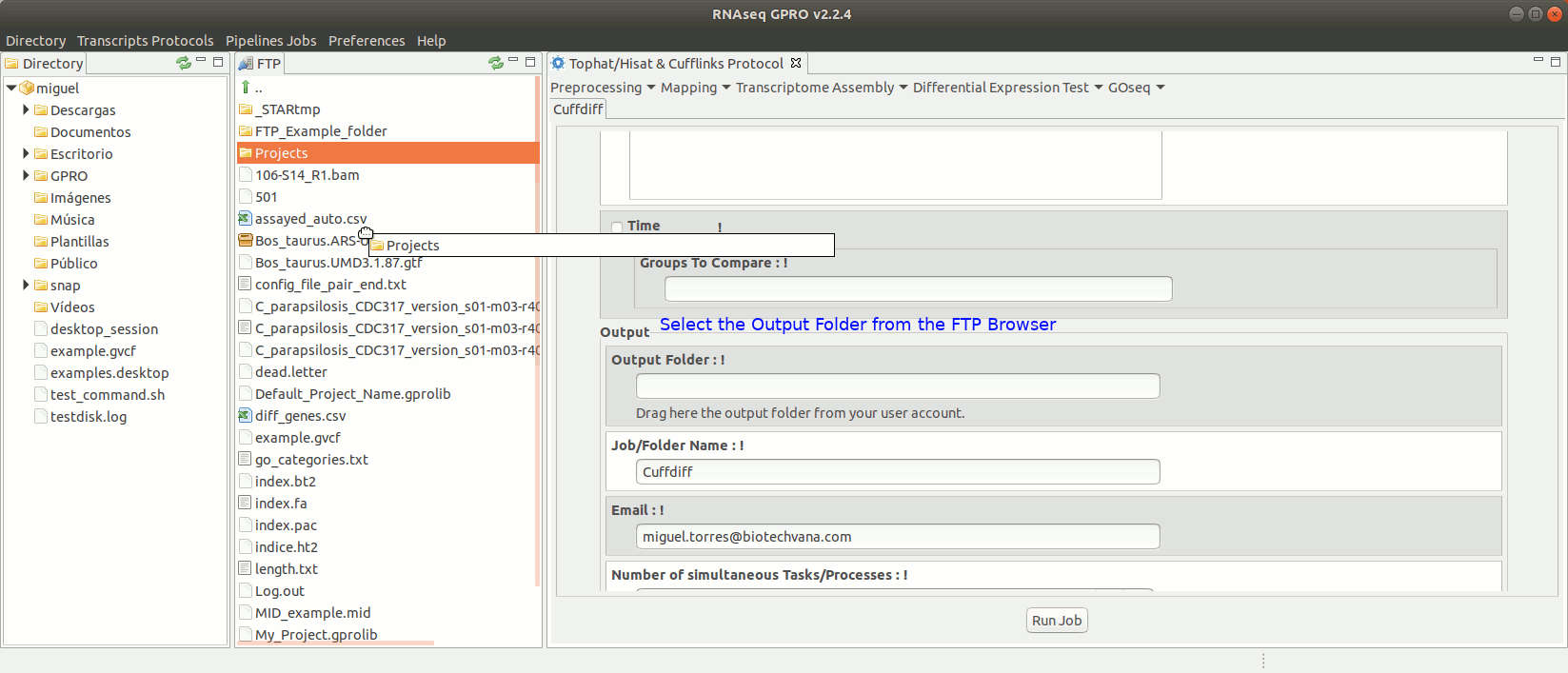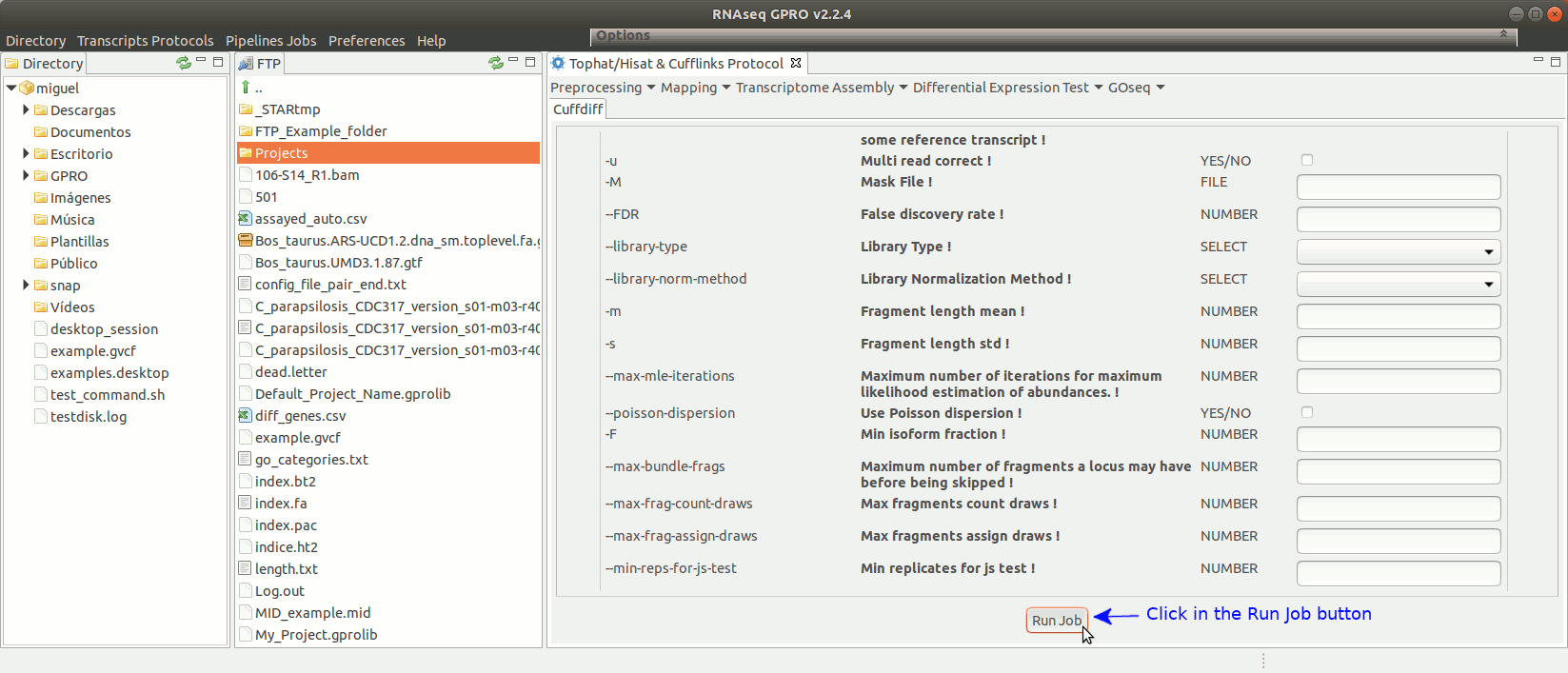
Figure 18: Using the GPRO interface for Cuffdiff.
- GoSeq: GoSeq
GOseq allows to work modes, auto and custom, that can be selected at the top of the ‘Input’ dialog in the GOseq interface. Through the auto mode, enrichment analyses can be based on natively supported genomes (that is, those listed in Ensembl) while the ‘custom’ option is used for non-native genomes and transcriptomes. In this case, the user must prepare the necessary input material according to the results found in the differential expression analysis.
To run GOseq go to[ GOseq→ GOeq ] and follow Fig. 19.
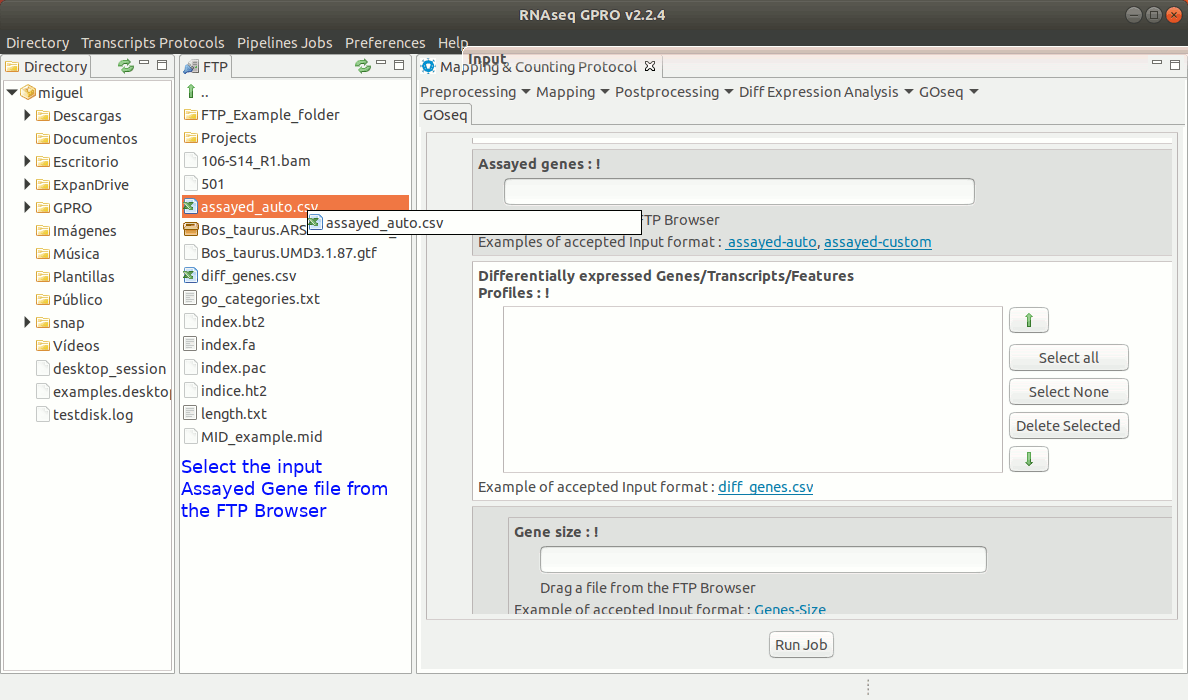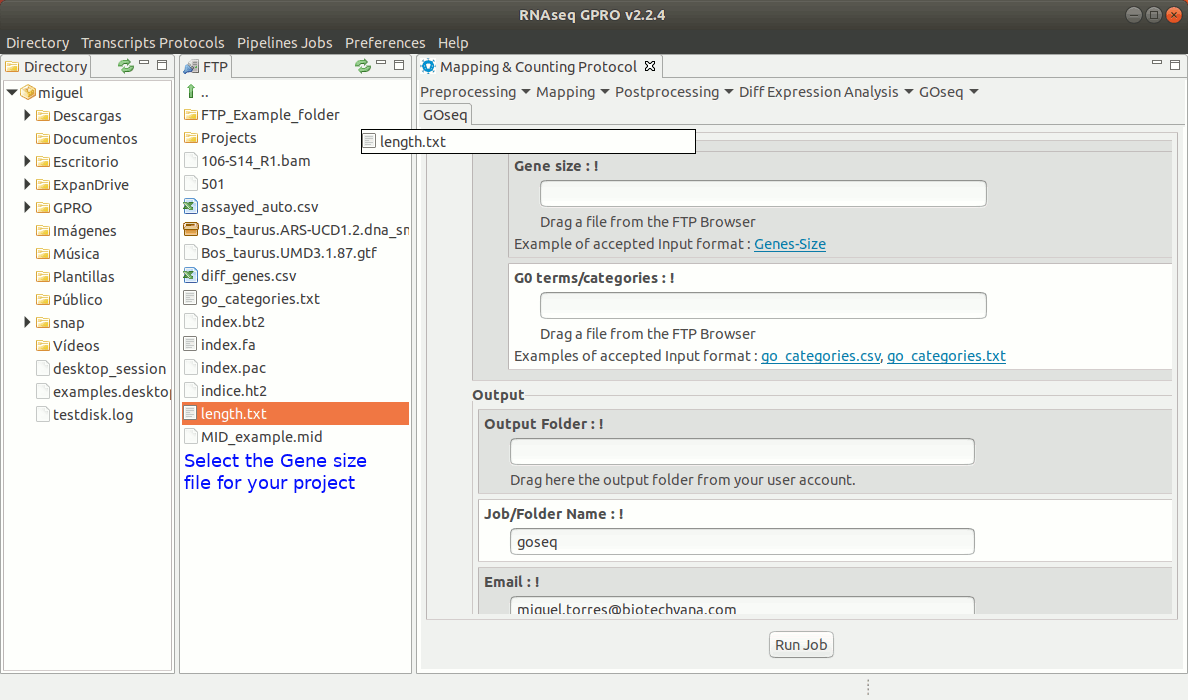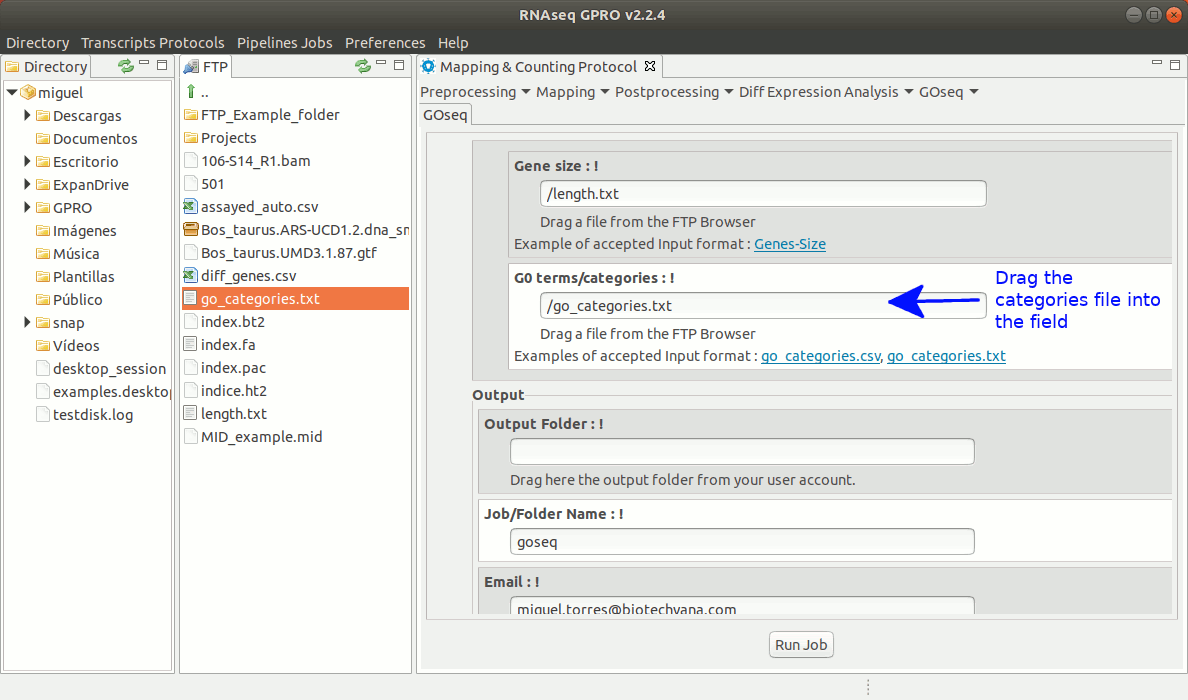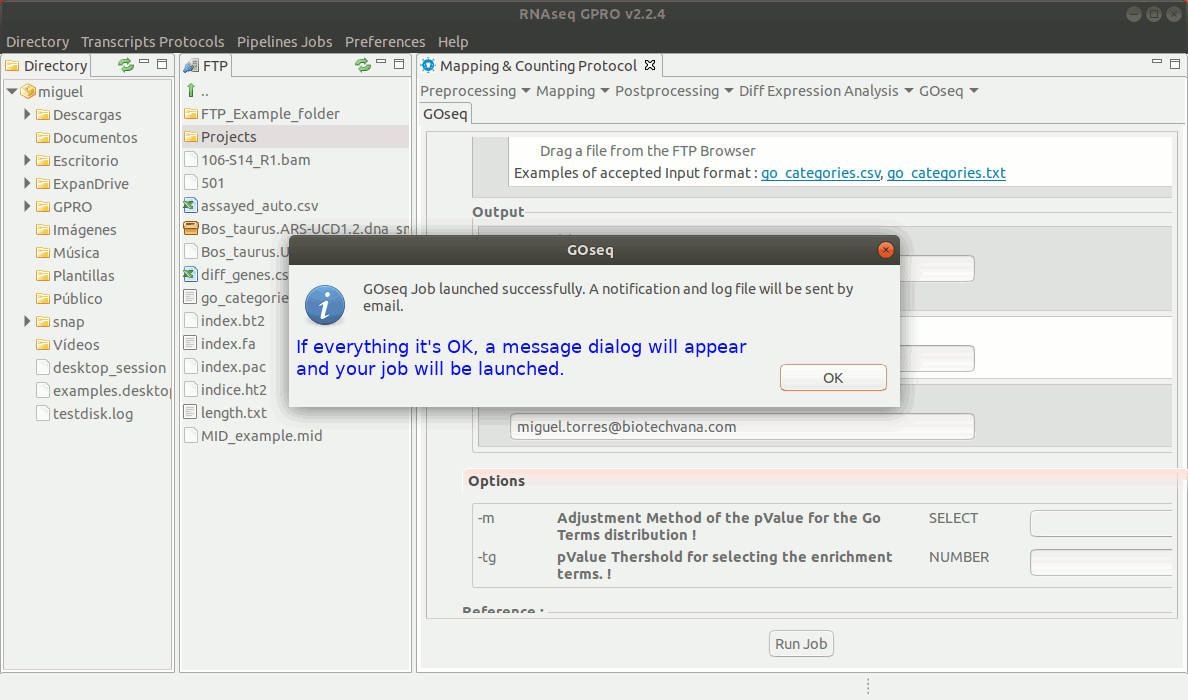
Figure 19: Using the GPRO interface for GOSeq.
[ Transcripts Protocols → Step-by-Step Mode → Mapping & Counting Protocol ]. Then a new submenu will appear in the workspace organizing the different steps that are required to perform an RNA-seq analysis without a reference, as follows [ Preprocessing -> Mapping -> Post processing -> Diff Expression Analysis -> Goseq ]
- Mapping: Bowtie2
Bowtie2 is an ultrafast and memory-efficient tool for the aligning of sequencing reads onto long reference sequences. For further details on this tool, see the Bowtie2 manual at "http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml.
To run Bowtie2 go to[ Mapping→ Bowtie2 ] and follow Fig. 20.
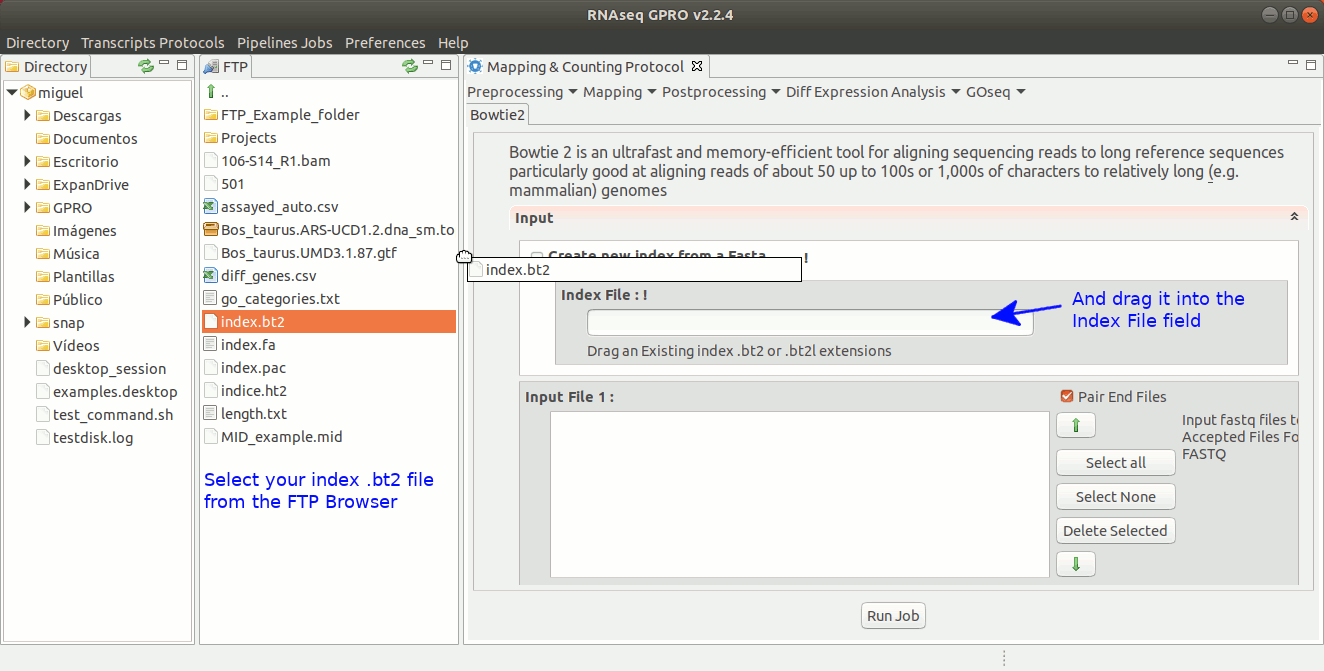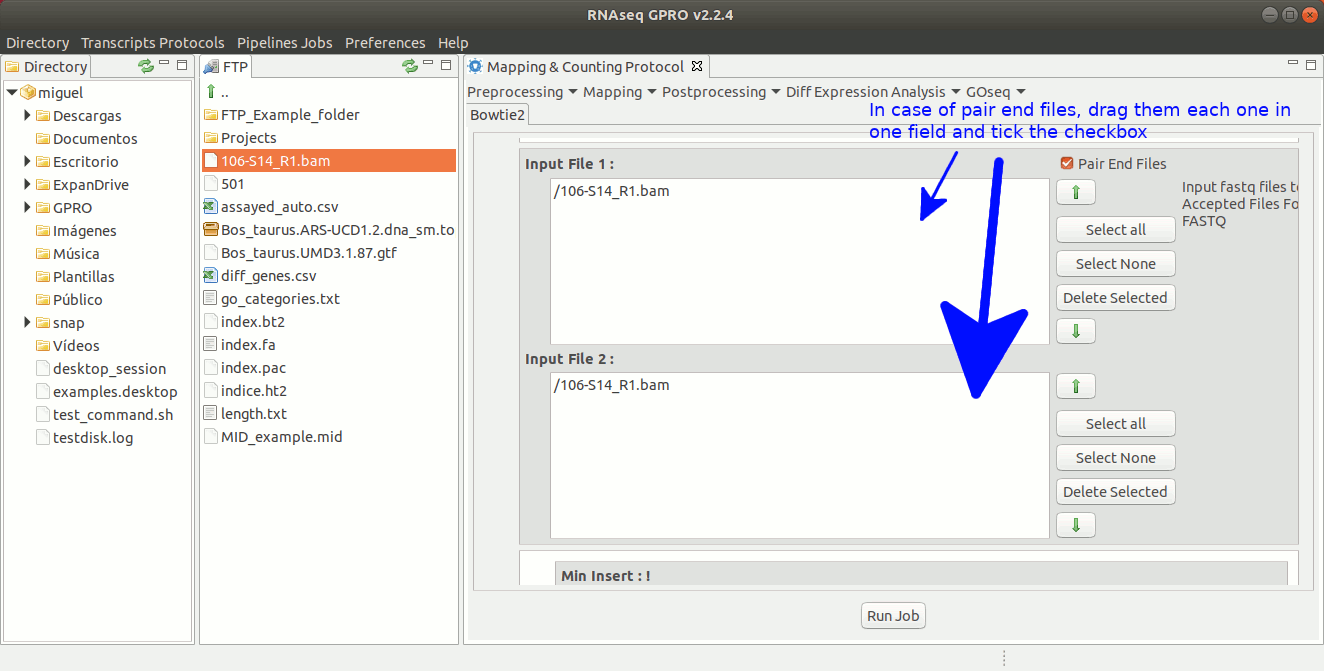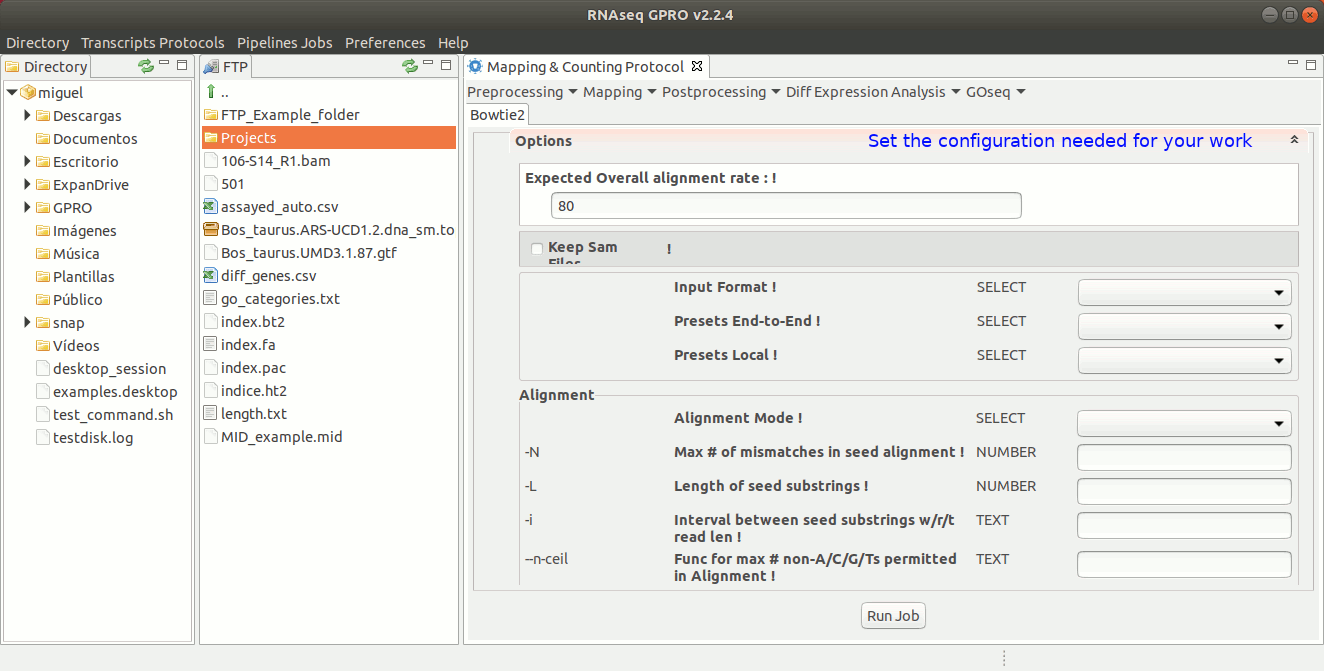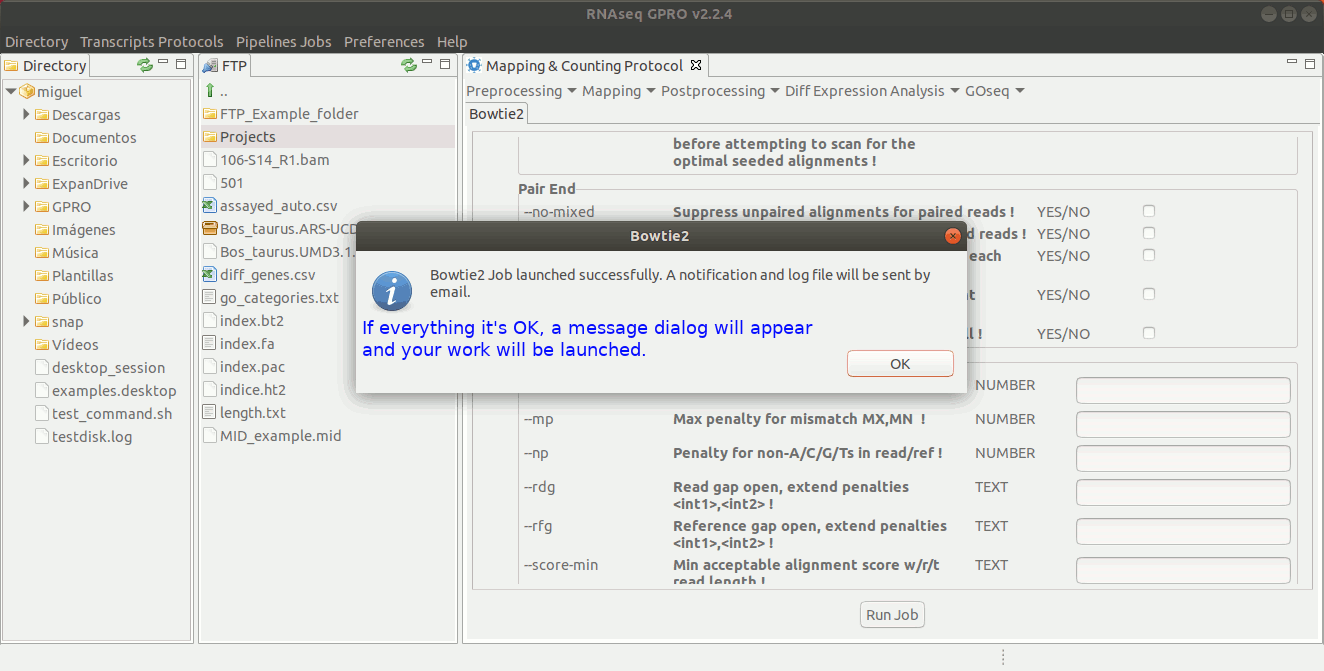
Figure 20: Using the GPRO interface for Bowtie2.
- Mapping : BWA
BWA is a software package for the mapping of low-divergent sequences against large reference genomes. It consists of three different algorithms, namely BWA-backtrack, BWA-SW and BWA-MEM. The first algorithm is specifically designed for Illumina sequence reads up to 100bp in length, while the other two are implemented for sequences ranging from 70bp to 1Mbp. BWA-MEM and BWA-SW share features such as long-read support and split alignment. BWA-MEM however is generally recommended for high-quality queries, as it is faster and more accurate. For more information, please refer to the BWA manual at http://bio-bwa.sourceforge.net/bwa.shtml”.
To run BWA go to [ Mapping → Bwa ] and follow Fig. 21.
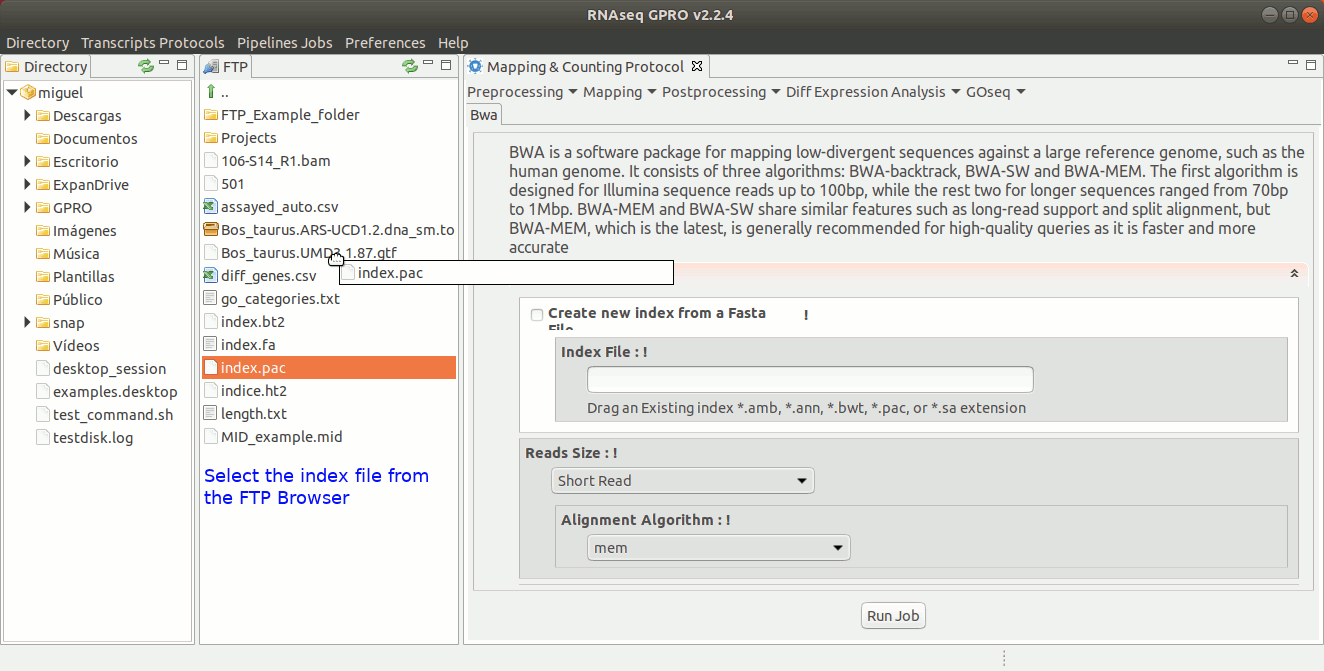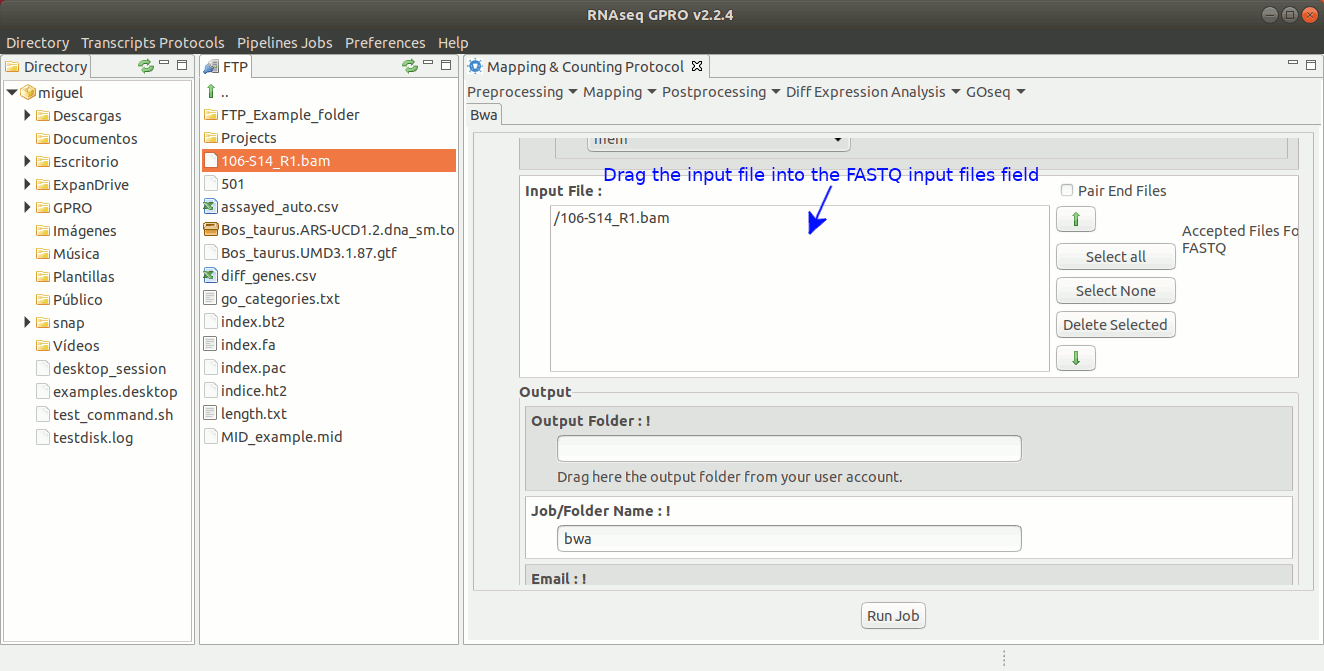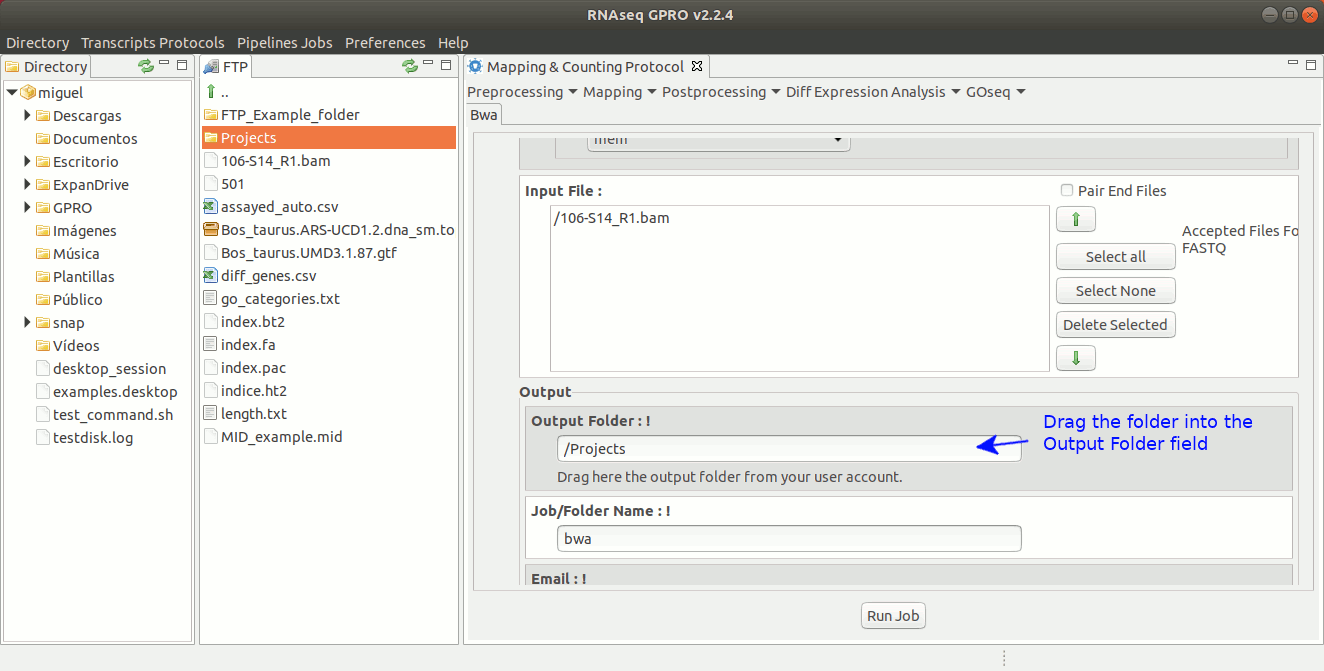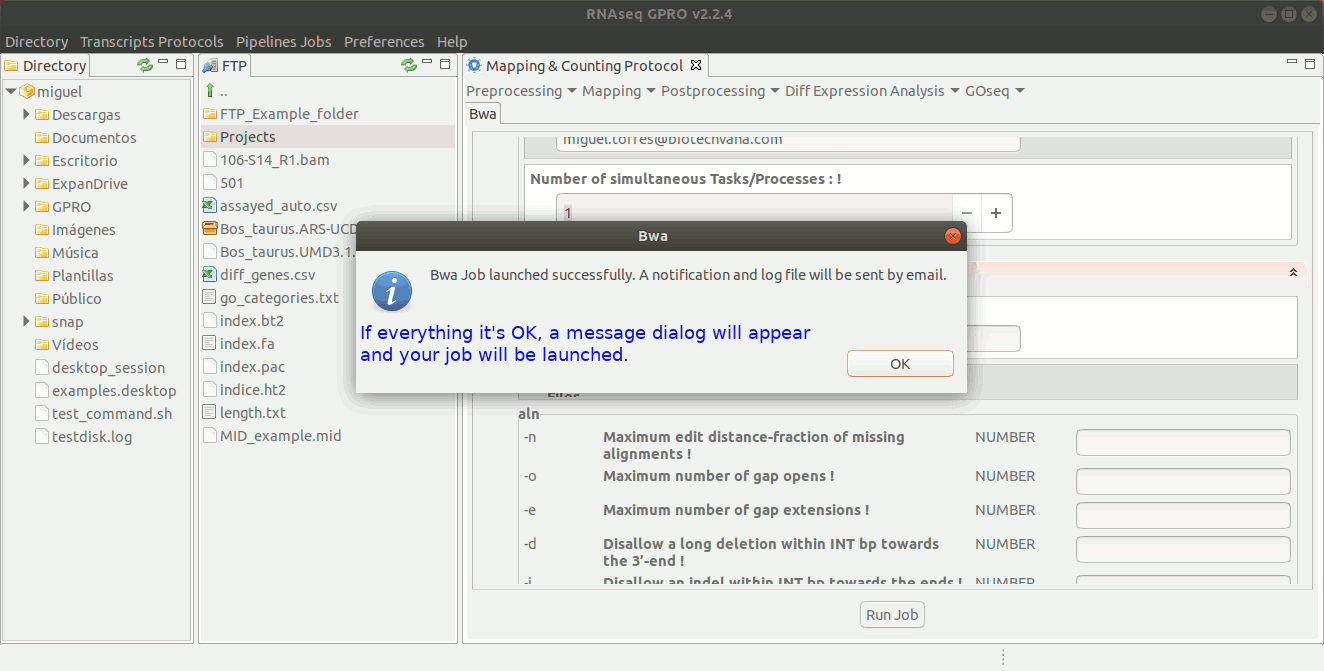
Figure 21: GPRO interface for BWA.
- Mapping : Hisat2
The interface provided for mapping with Hisat2 is the same as the one described for the Tophat & Cufflinks protocol. For more details, please refer to Section 2.2.2 of this manual.- Postprocessing : Corset
Corset has been traditionally used to obtain gene-level counts of de novo-obtained transcriptome assemblies. To do so, Corset uses the reads that have been mapped to the transcriptome to hierarchically cluster them according to the proportion of shared reads and their expression patterns. Subsequently, the clusters and gene-level counts for each sample are reported. The output generated by Corset is the input that will be required by the counting-based tools EdgeR and DESeq to later perform the differential expression analysis. For more information, please refer to the Corset manual at https://github.com/Oshlack/Corset/wiki/InstallingRunningUsage.
To run Corset go to [ Postprocessing → Corset ] and follow Fig. 22.
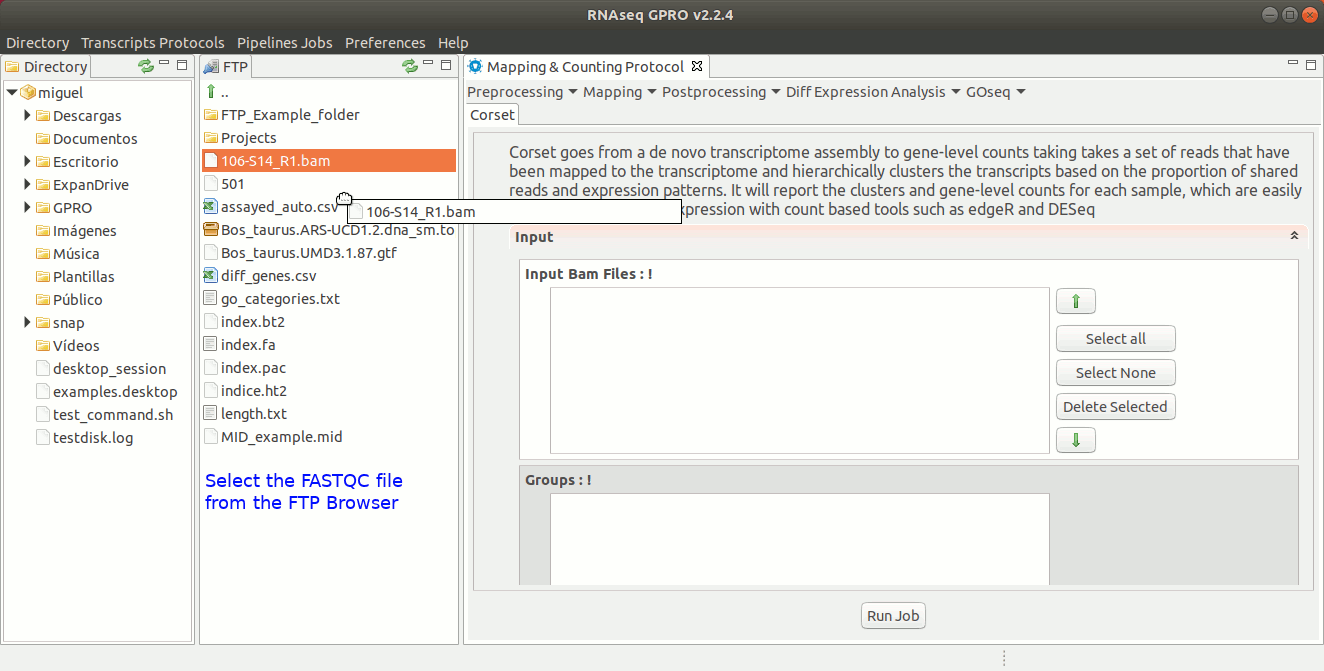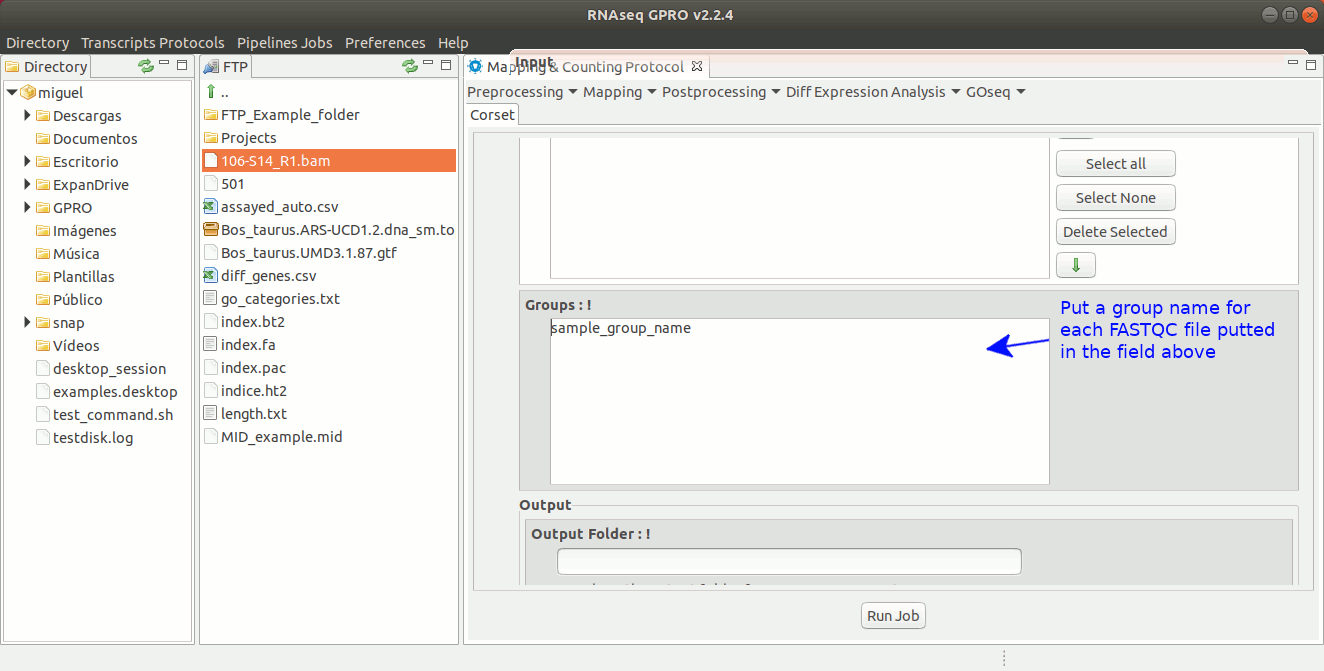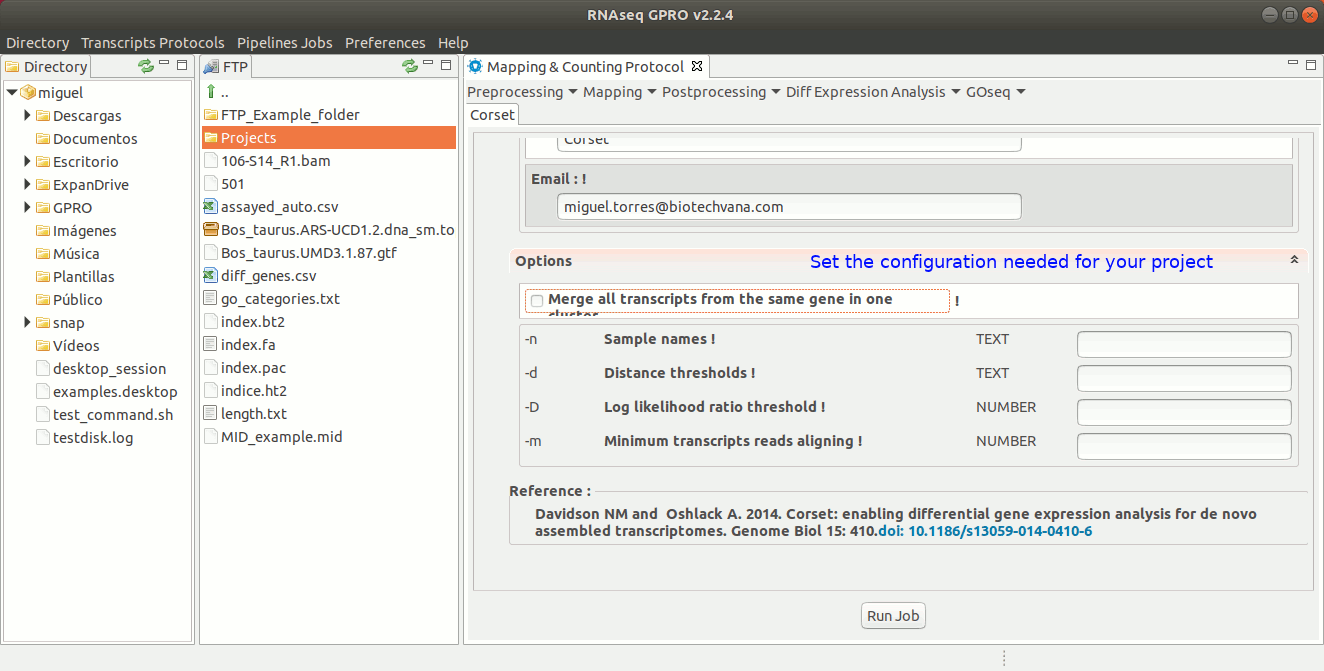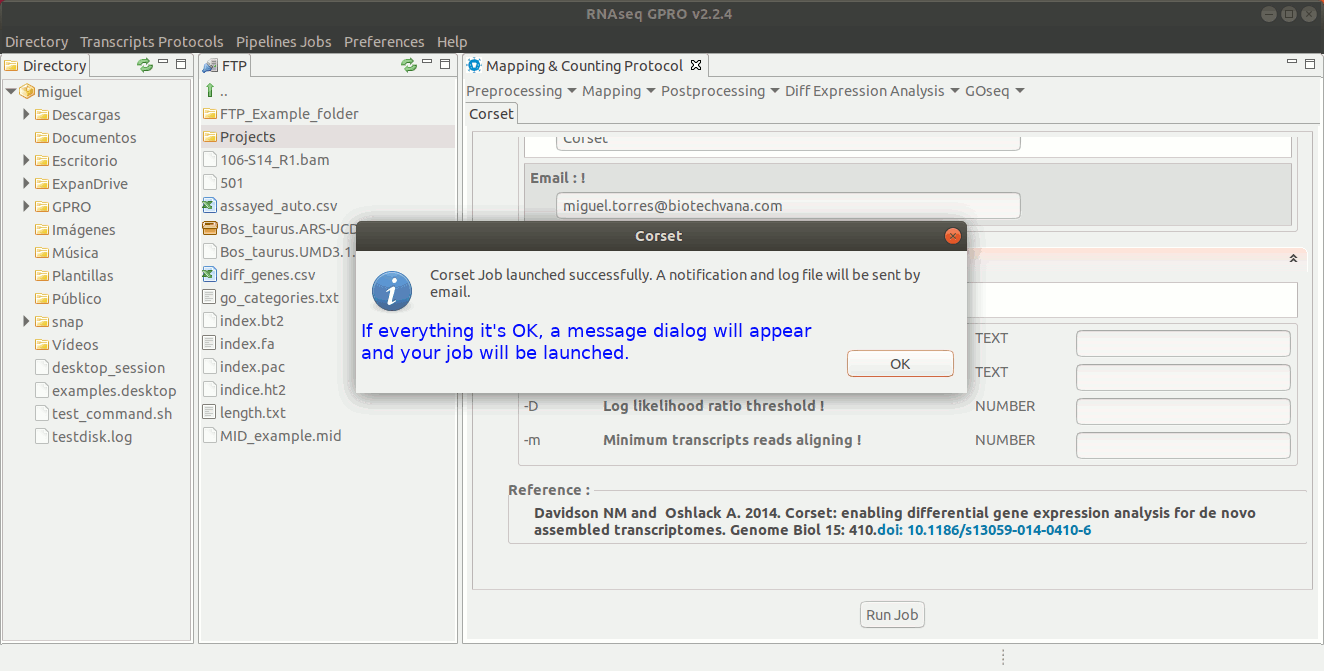
Figure 22: Using the GPRO interface for Corset.
- Postprocessing : Htseq
For this post-processing option, RNASeq makes use of the ‘Htseq-count’ option of the Htseq tool. This tool counts those reads that are mapped to genomic features (exons, genes, etc) reporting a counts file at the selected feature-level (in RNA-Seq, typically genes). The output generated by Htseq-count will be the input required by the tools EdgeR and DESeq to perform the differential gene expression analysis. For more information, please refer to the Htseq manual at https://htseq.readthedocs.io/en/release_0.9.1/count.html .[ Postprocessing→ Htseq ] and follow Fig. 23.
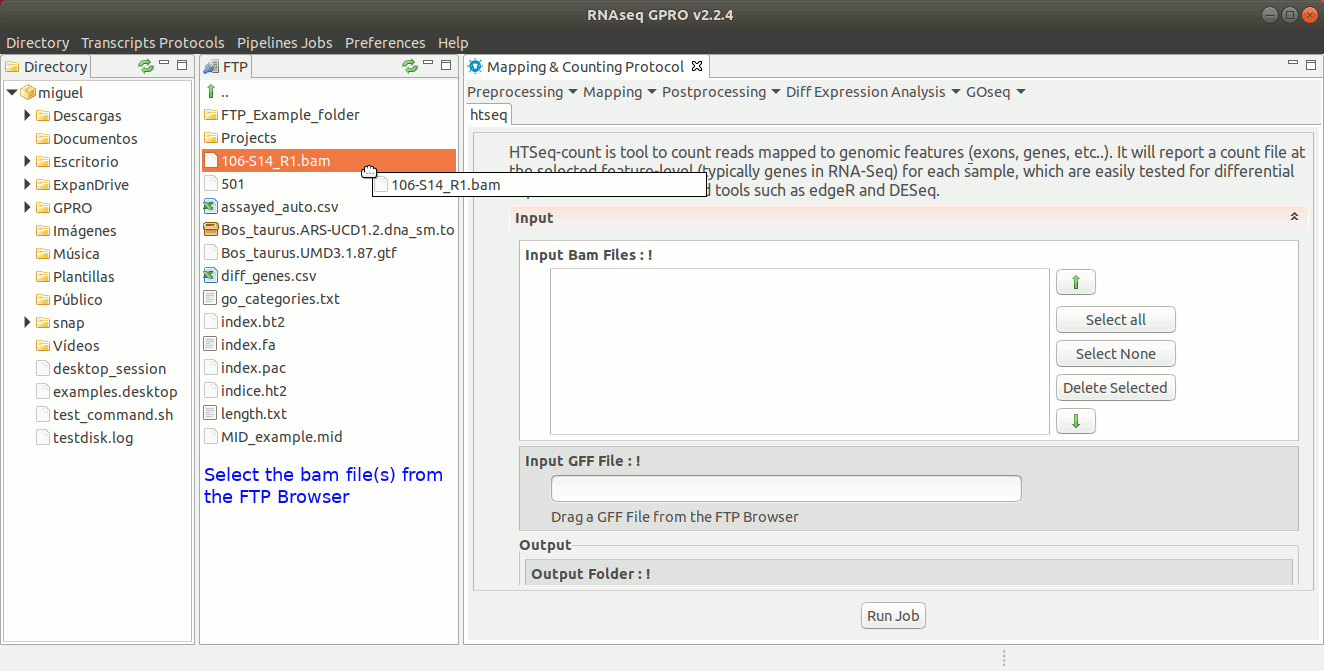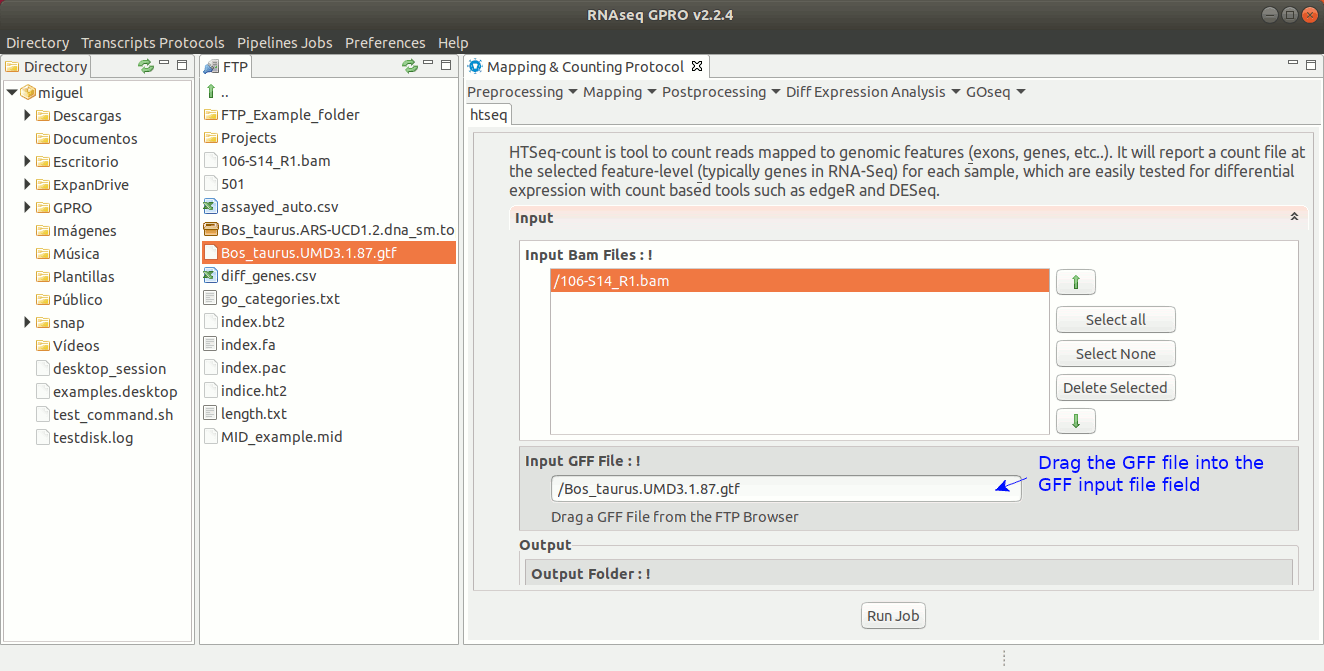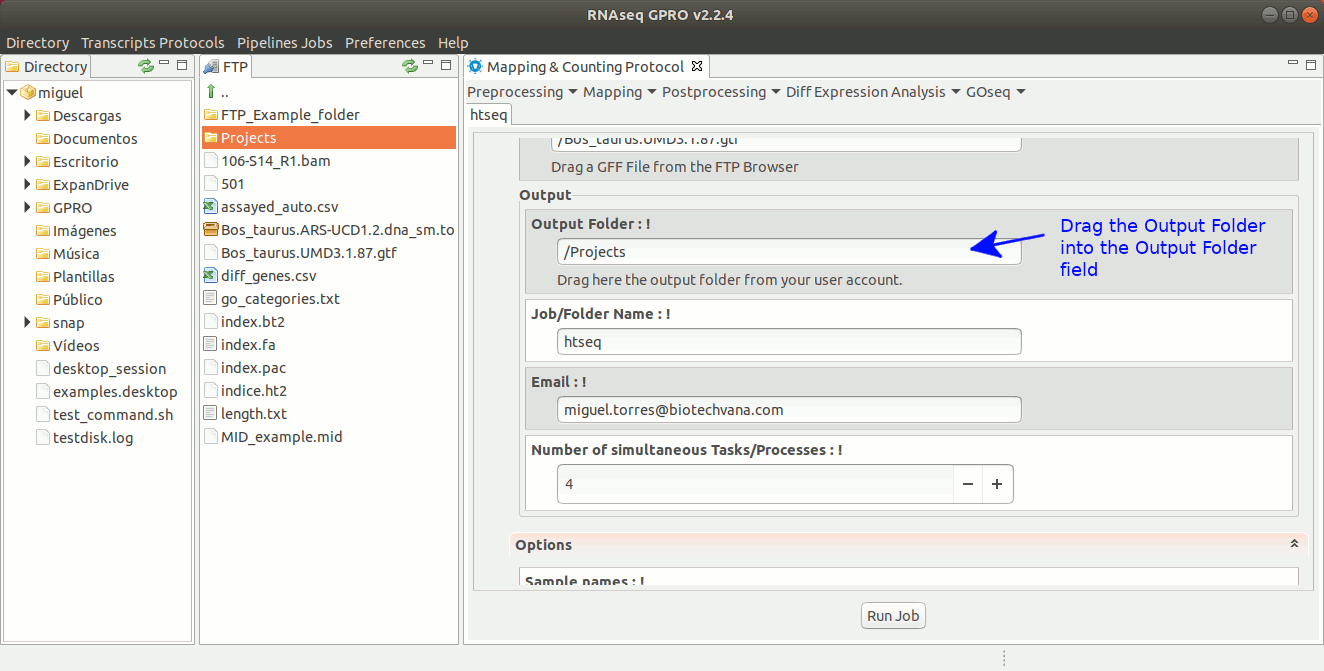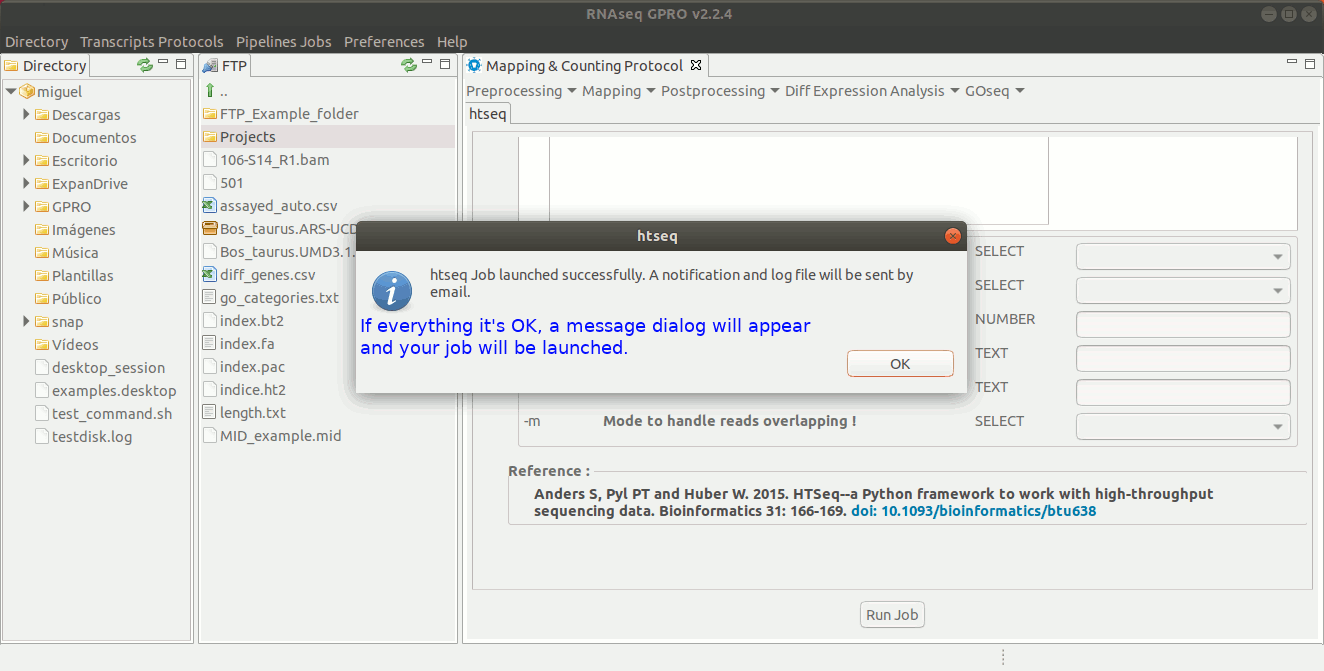
Figure 23: Using the GPRO interface for HtSeq.
- Diff Expression Analyses : DESeq
DESeq estimates variance-mean dependences in sequencing read count data to then test for differential gene expression using a model that is based on the negative binomial distribution. For more information please refer to the DESeq manual at https://bioconductor.org/packages/3.8/bioc/html/DESeq.html.
To run DESeq go to [ Diff Expression Analysis → DESeq ] and follow Fig. 24.
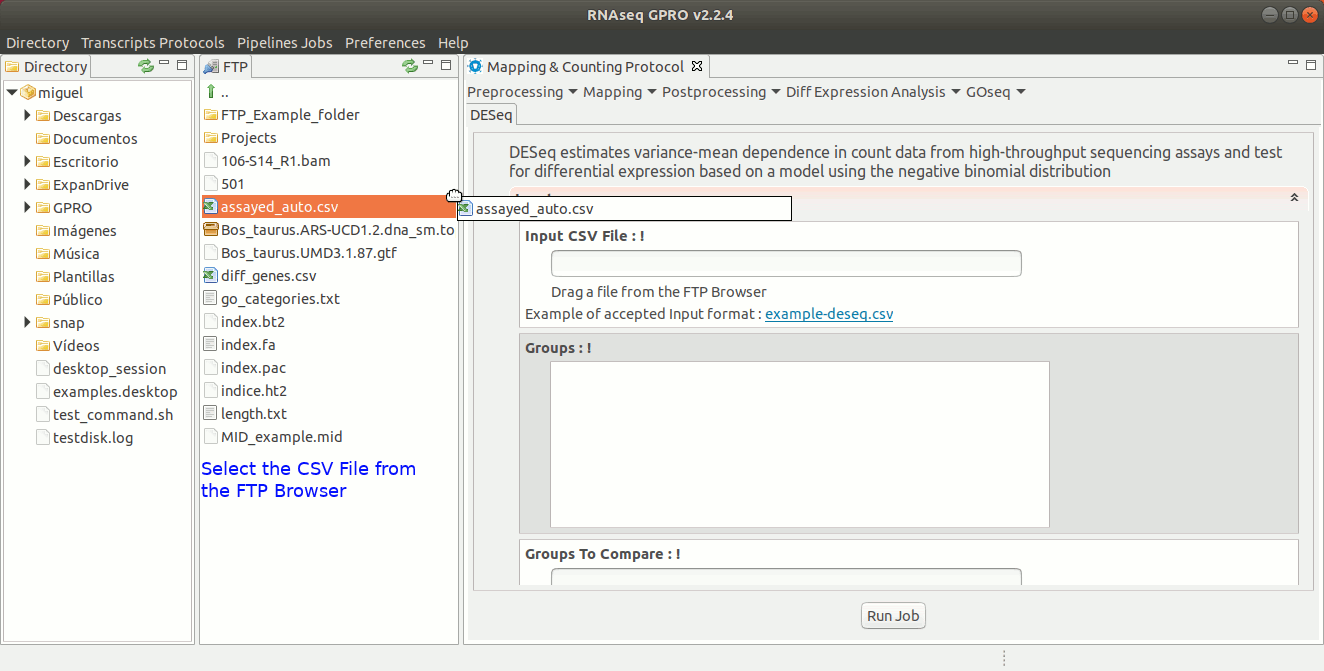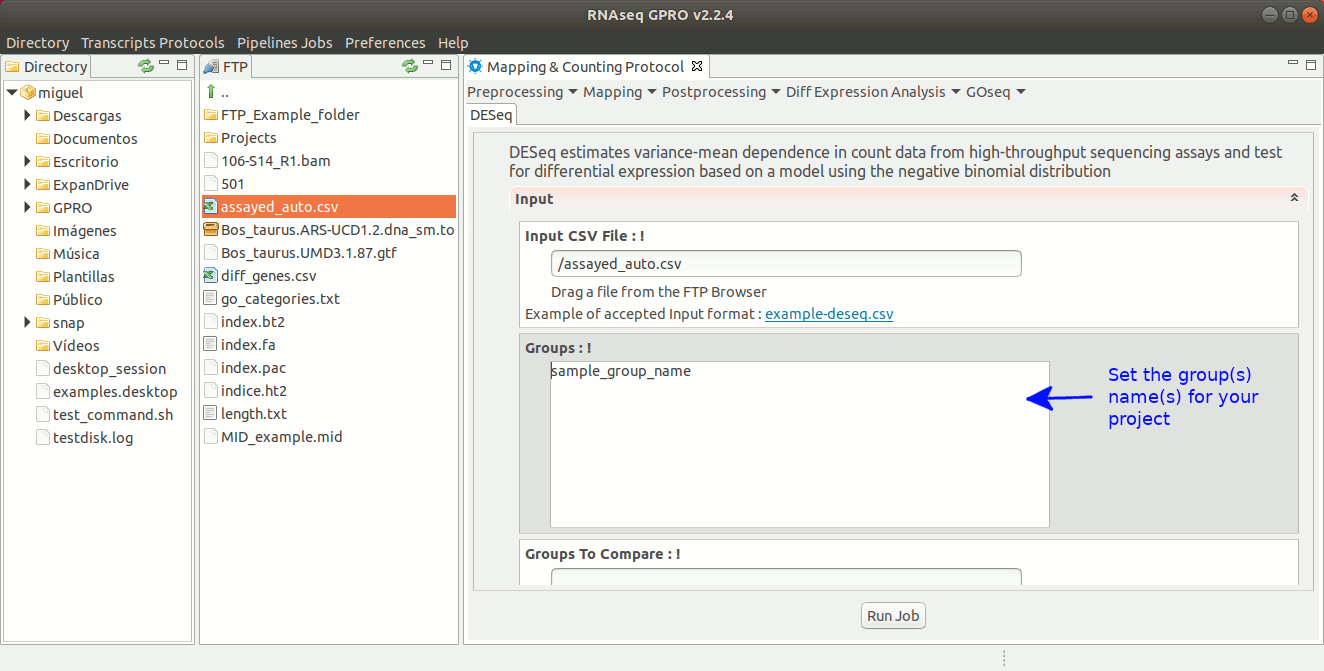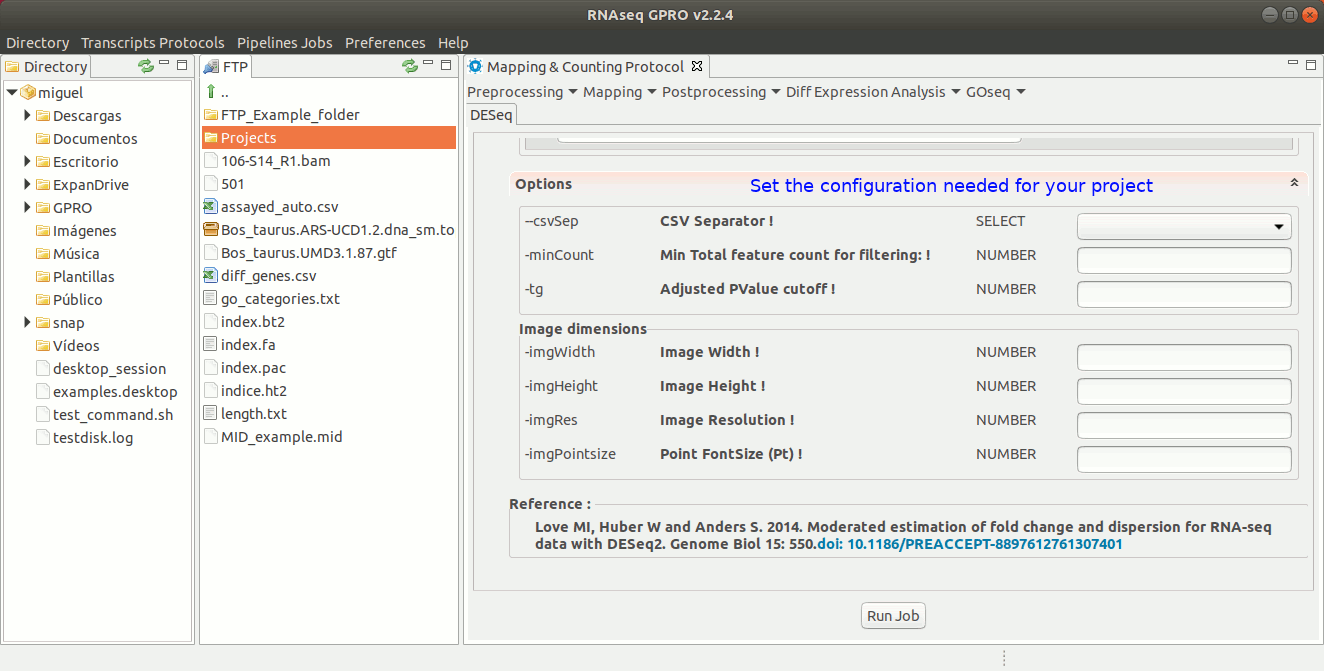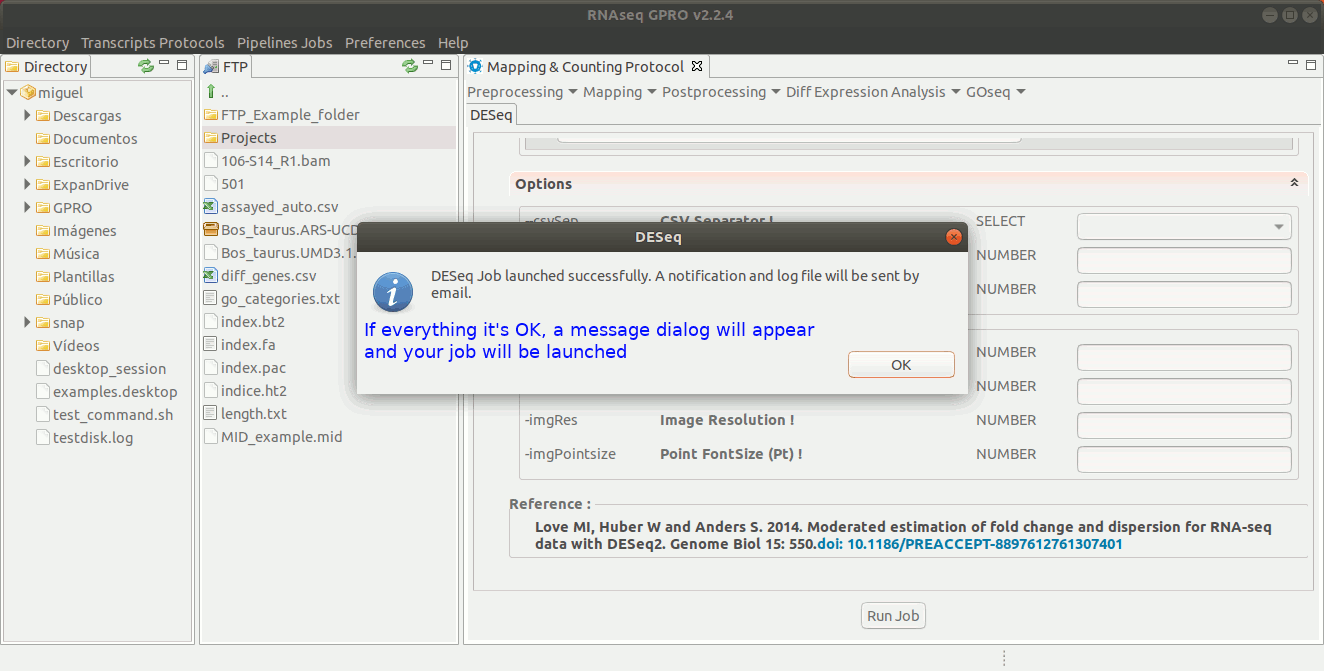
Figure 24: Using the GPRO interface for DeSeq.
Diff Expression Analyses : EdgeR
EdgeR performs differential gene expression analyses using an array of statistical methods that are based on negative binomial distributions including empirical Bayes estimation, exact tests, generalized linear models and quasi-likelihood tests. For more information please refer to the EdgeR manual at https://bioconductor.org/packages/release/bioc/html/edgeR.html.
To run EdgeR go to [ Diff Expression Analysis → EdgeR ] and follow Fig. 25.
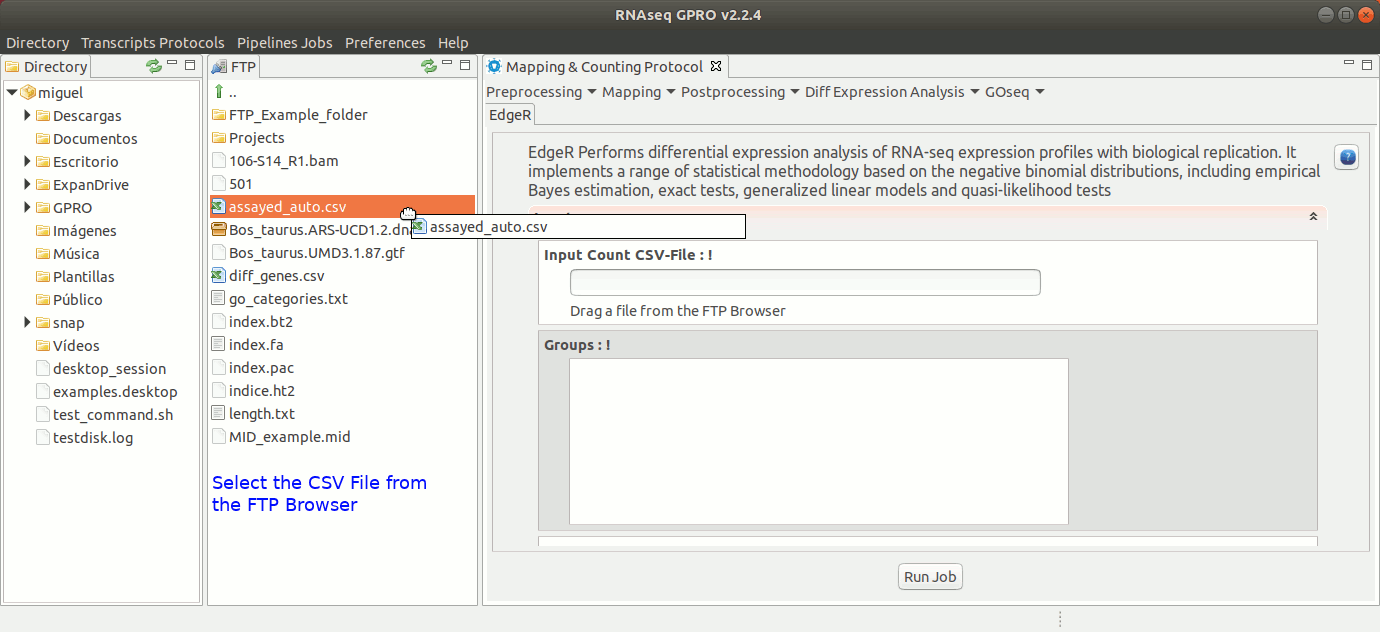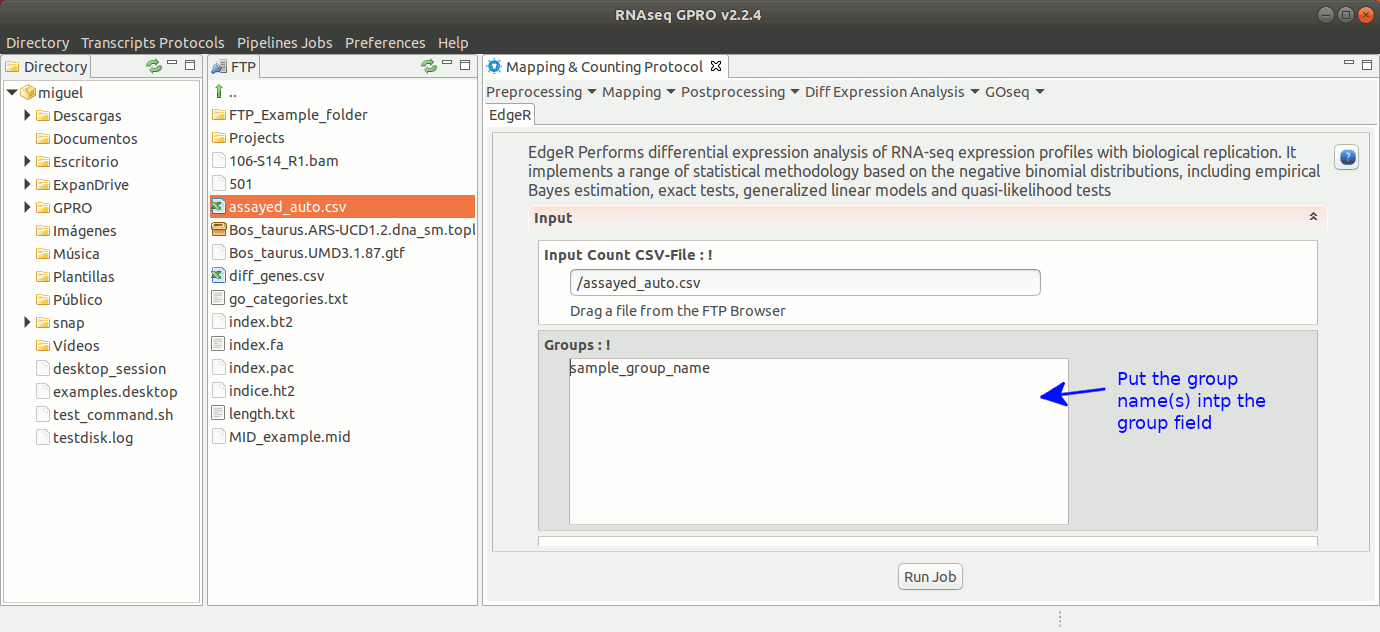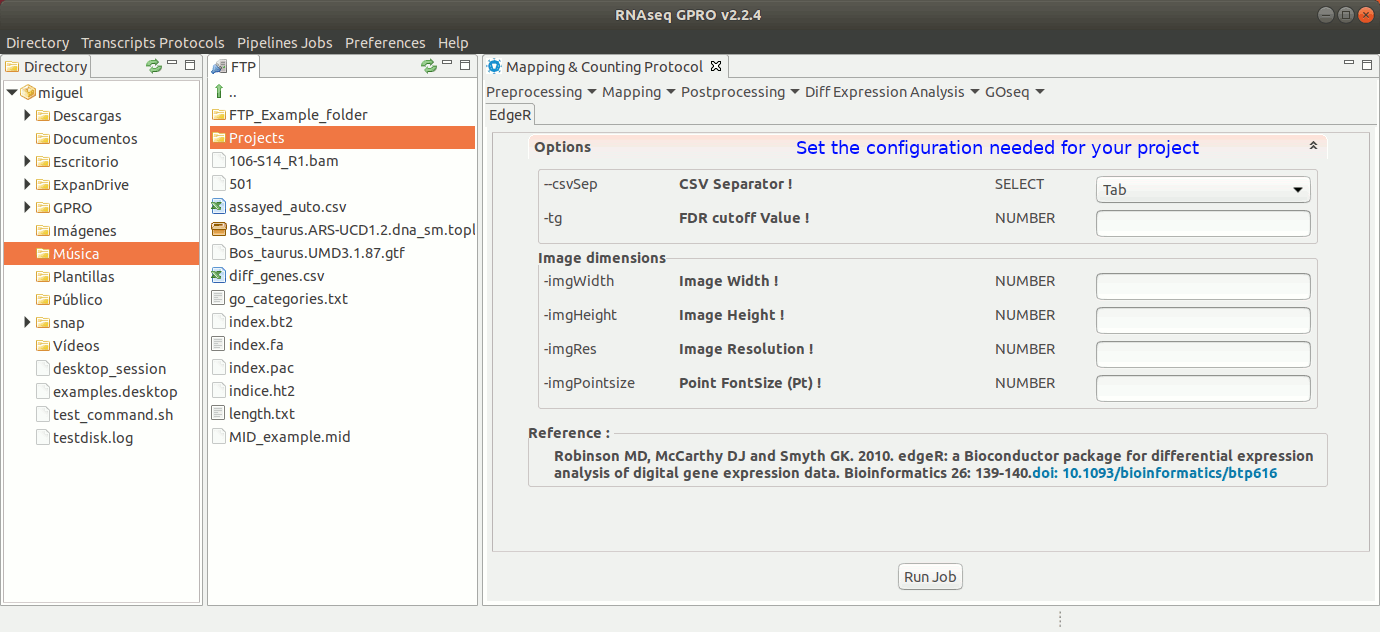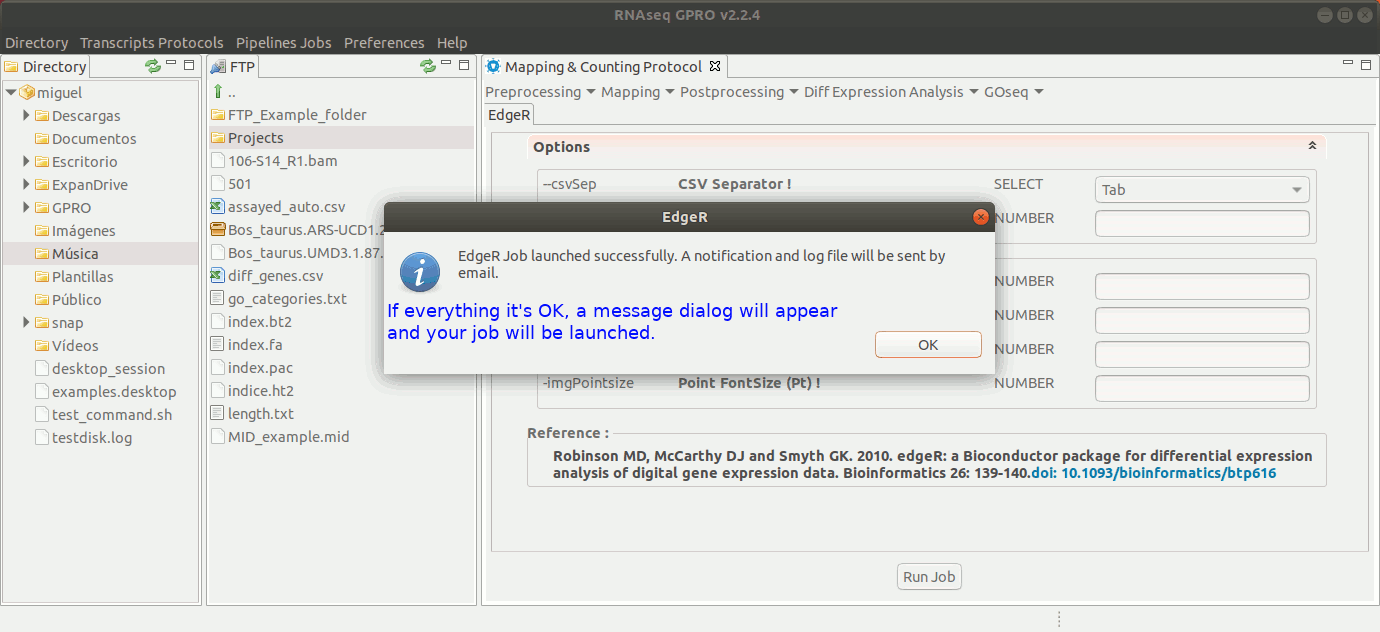
Figure 25: Using the GPRO interface for EdgeR.







